Das Makrophagenaktivierungssyndrom (MAS) beziehungsweise die Hämophagozytische Lymphohistiozytose ist eine seltene Erkrankung, deren Prognose durch hohe Letalität gekennzeichnet ist. Man unterscheidet primäre Formen von sekundären, reaktiven Formen, die auf Virus- oder Bakterieninfektionen, hämatoonkologische Erkrankungen und bestimmte Autoimmunerkrankungen zurückgehen können. In diesem Artikel befassen wir uns mit den sekundären Formen des MAS.
Fallbeispiel
Bei dem Patienten – einem 24-jährigen Asylsuchenden aus Eritrea, der sich seit zwei Monaten in der Schweiz befindet und sich sonst guter allgemeiner Gesundheit erfreut – sind Fieberzustände mit Spitzen von bis zu 40 °C zu beobachten, die sich seit sechs Wochen entwickeln und mit Kopf- und Muskelschmerzen einhergehen. Aus diesem Grund hat er bereits ein anderes Spital aufgesucht. Umfassende serologische Untersuchungen (Bartonella, Brucella, Coxiella, Salmonella, Syphilis, HIV, HCV, HBV, Dengue, Leptospirose) erbrachten ebenso negative Ergebnisse wie ein Quantiferon-Test und ein «dicker Tropfen» zum Malarianachweis. In der transthorakalen Echokardiografie sind keine Anzeichen einer Endokarditis feststellbar.
Die klinische Untersuchung ergibt eine Hepatosplenomegalie ohne sonstige Auffälligkeiten. Die labormedizinischen Tests zeigen ein ausgeprägtes Entzündungssyndrom, eine Panzytopenie, gestörte Hämolyseparameter (bei negativem Coombs-Test) sowie eine Leberzytolyse (ohne assoziierte Cholestase). Aufgrund des Anfangsverdachts auf Typhus wird eine Antibiotikatherapie mit Ceftriaxon begonnen, aber bald wieder abgesetzt, da die bakteriologische Untersuchung von Blut- und Stuhlproben negativ verläuft. Eine Thorax-Abdomen-Becken-Computertomografie ist unauffällig, abgesehen von der Hepatomegalie und einer erheblichen Splenomegalie. Der Krankheitsverlauf ist gekennzeichnet durch einen andauernden Fieberzustand und die Verschlimmerung der genannten labormedizinischen Befunde. Die Elektrophorese der Serumproteine weist auf Störungen hin, die mit einem chronischen Entzündungszustand vereinbar sind (erhöhte Konzentration der Alpha-1- und Alpha-2-Globuline, Hypoalbuminämie, verringerte Konzentration der Beta-1-Globuline).
Angesichts des klinischen Bildes wird ein Makrophagenaktivierungssyndrom (MAS) diagnostiziert, nachdem eine erhebliche Hyperferritinämie (11900 µg/l) und eine Hypertriglyzeridämie (3,35 mmol/l) nachgewiesen wurden. Das Hämatomyelogramm weist auf eine Hämophagozytose und ein trilinear hyperzelluläres Knochenmark hin. Sechs der acht HLH-2004-Kriterien (siehe unten) sind erfüllt und der HScore beträgt 299 (Wahrscheinlichkeit eines MAS über 99%): Dies bestätigt die Diagnose. Die Abklärung der Ätiologie wird durch einen Test auf viszerale Leishmaniose vervollständigt, der bisher noch nicht durchgeführt wurde. Die direkte Blutuntersuchung liefert keine Hinweise, die serologischen Tests sind jedoch ebenso positiv wie die PCR-Analyse der Hämatomyelogramm-Probe. Als endgültige Diagnose wird folglich eine viszerale Leishmaniose mit sekundärem MAS festgestellt.
Wir weisen darauf hin, dass in dem beschriebenen Fall die Leishmaniose nicht die übliche pathognomonische Trias aufweist: Lediglich die ausgeprägte Splenomegalie liegt vor, nicht jedoch die polyklonale Hypergammaglobulinämie und der positive Coombs-Test. Darüber hinaus wurde die Leishmaniose nicht mithilfe der gemeinhin beschriebenen Methode gestellt (Leishmanien-Nachweis im Makrophagenzytoplasma mittels PAS-Reaktion [«Periodic acid-Schiff»]), sondern durch das PCR-Verfahren. Infolge einer Behandlung mit Amphotericin B (fünf Tage lang und an den Tagen 7 und 14) klingen die klinischen und labormedizinischen Symptome rasch ab.
Einleitung
Das MAS wurde erstmals 1939 von Scott und Robb-Smith unter der Bezeichnung «medullär histiozytäre Retikulose» beschrieben [1] und ist gekennzeichnet durch ein schwerwiegendes, durch die Deregulierung der zytotoxischen Zellreaktion ausgelöstes Entzündungssyndrom. Primäre (genetische) Formen treten vor allem in der pädiatrischen Population auf, sekundäre (reaktive) Formen sind dagegen in jeder Altersgruppe zu finden. Der mögliche schwere Verlauf rechtfertigt aggressive Diagnose- und Therapiemassnahmen. Da sich der Grossteil der Prospektiv- und Interventionsstudien bisher mit dem primären MAS beschäftigte, sind die Diagnosekriterien und die Therapie des sekundären MAS noch ungenügend belegt. Die Krankheit ist selten, doch ihre Prävalenz ist wohl unterschätzt [2]. In Japan wird die jährliche Inzidenz auf einen Fall pro 800000 Einwohner geschätzt [3]. Für das sekundäre MAS im Rahmen einer Hämopathie wurden jährliche Inzidenzen von 3,6 Fällen pro 1000000 Erwachsene berichtet [4]; für Formen, die als Komplikation einer Infektion auftreten, betrug die Inzidenz 0,9 Fälle pro 1 000000 Erwachsene [5]. Ein virusassoziiertes MAS scheint in jeder Altersgruppe auftreten zu können [6]. Im Hinblick auf das Geschlecht besteht anscheinend kein Unterschied.
Pathophysiologie
Mithilfe experimenteller Modelle zur Untersuchung des primären MAS wurden in jüngerer Vergangenheit Fortschritte beim Verständnis der Pathophysiologie des sekundären MAS erzielt.
NK-Zellen (natürliche Killerzellen) sind Bestandteil des angeborenen Immunsystems und verfügen über eine spontane Zytotoxizität. Sie können Tumorzellen oder infizierte Zellen eliminieren und gleichzeitig Zytokine freisetzen, welche die Reaktion der B- und T-Lymphozyten steuern. Werden sie aktiviert, sezernieren sie zytotoxische Granula, die Perforin [7] und Granzyme enthalten. Dadurch kommt es zur Perforation der Zellmembran und zur Apoptose der Zielzelle. Den zytotoxischen T-Lymphozyten kommt eine ähnliche Funktion wie den NK-Zellen zu. Voraussetzung dafür ist, dass sie ein Peptidantigen erkannt haben, das durch antigenpräsentierende Zellen in Verbindung mit Molekülen des Haupthistokompatibilitätskomplexes vorgezeigt wird. Im Rahmen einer normalen Immunantwort werden die NK-Zellen, die T-Lymphozyten und Histiozyten (Fähigkeit zur Phagozytose und zur Antigenpräsentation) durch proinflammatorische Zytokine aktiviert, um das Zielantigen zu zerstören. Sobald das Zielantigen verschwunden ist, wird die Immunantwort beendet.
Beim primären MAS ist eine verringerte Zytotoxizität der T-Lymphozyten und/oder NK-Zellen zu beobachten, die auf einen Mangel an Perforin oder einen nicht funktionellen Granula-Exozytose-Weg zurückzuführen ist [8]. Infolge der ungenügenden Eliminierung der Antigene kommt es zu einer Proliferation der T-Lymphozyten und NK-Zellen und somit zu einer massiven Produktion proinflammatorischer Zytokine (TNF-α, IFN-γ, IL-1β, IL-6, IL-8, IL-12, IL-18): Man spricht dann von einem Zytokinsturm. Die Freisetzung der Zytokine verursacht die Aktivierung der Makrophagen und dadurch die klinischen, labormedizinischen und zytologischen Manifestationen. Bei den reaktiven Formen des MAS kann die anhaltende Aktivierung der T-Lymphozyten und NK-Zellen durch eine vorbestehende Immundepression bedingt sein (HIV-Infektion, Immunsuppression, hämatologische Krankheit), die eine angemessene Immunantwort und die Eliminierung des Antigens verhindert; oder aber durch einen externen Reiz (etwa eine Virusinfektion), der die Toll-like-Rezeptoren 9 (TLR-9) oder den MyD88-abhängigen Signalweg über einen längeren Zeitraum stimuliert und die antigenpräsentierenden Zellen aktiviert [9, 10].
Toll-like-Rezeptoren und MyD88-abhängiger Signalweg
Toll-like-Rezeptoren (TLR) sind Transmembranrezeptoren, die an antigenpräsentierenden Zellen (dendritische Zellen, Makrophagen, Monozyten, B-Lymphozyten) nachweisbar sind. Sie erkennen pathogenassoziierte Molekülmuster (Proteine, Nukleinsäuren) und spielen bei der angeborenen Immunität eine zentrale Rolle. Beim Menschen sind 13 verschiedene TLR bekannt. Die Aktivierung eines TLR löst intrazellulär eine Signalkaskade aus und folglich die Transkription bestimmter Gene, denen eine Funktion bei der Immunantwort zukommt.
Das intrazelluläre Protein MyD88 («myeloid differenciation primary response 88») beeinflusst den gleichnamigen Signalweg und ermöglicht – in Verbindung mit anderen Molekülen – die Übertragung des Signals zwischen den TLR und den Transkriptionsfaktoren, die ihrerseits die Aktivität bestimmter Gene lenken. So können die Immunantwort und die Entzündungsreaktion gesteuert werden. Eine anhaltende Stimulierung des TLR-9 oder des MyD88-abhängigen Signalwegs dereguliert die Immunantwort und die Entzündungsreaktion und trägt so zur Entstehung eines MAS bei [10, 11].
Ätiologie
Ein MAS auf eine bestimmte Infektion zurückzuführen, ist nicht immer einfach, da Infektionen als Komplikation von Krankheiten auftreten können, die selbst als Ursache des MAS infrage kommen (etwa HIV-Infektion, Lymphom, Lupus). Mehrere potenziell MAS-verursachende Pathologien können assoziiert sein und die Feststellung der Ätiologie erschweren. In diesem Falle können wir prädisponierende und auslösende Faktoren unterscheiden (Tab. 1).
Tabelle 1: Ursachen des Makrophagenaktivierungssyndroms.
Die Ursachen lassen sich – nach Häufigkeit gereiht – in infektiöse, hämatoonkologische und autoimmune Ätiologien einteilen. Ihre Prävalenz ist geografisch unterschiedlich.
Infektionsbedingtes MAS
Infektionen sind vermutlich die häufigste Ursache für ein MAS [11]. 62% der sekundären MAS sind auf eine Virusinfektion zurückzuführen, sei es eine Erstinfektion bei einem gesunden Erwachsenen oder eine Reaktivierung bei einem Patienten mit Immundepression [12–14]. Überwiegend handelt es sich dabei um Herpesviren [2], besonders um das Epstein-Barr-Virus (EBV) und das Zytomegalie-Virus (CMV). Häufig werden auch Adenoviren, das Hepatitis-A-Virus, das Parvovirus B19 und Influenza-A-Viren (H1N1 und H5N1) nachgewiesen [15]. Eine Infektion mit dem Humanen Immundefizienz-Virus (HIV) kann – allein oder im Rahmen einer opportunistischen Infektion – mit einem MAS einhergehen [15].
Ein MAS kann auch als Komplikation jeder schweren Bakterieninfektion auftreten, vor allem wenn sie durch intrazelluläre Keime ausgelöst ist (Rickettsiose, Q-Fieber, Brucellose) [11]. Auch infolge einer durch pyogene Erreger verursachten Infektion kann ein MAS auftreten [16]. Als klassische Ursache gilt Tuberkulose. In 9% der sekundären Formen sind bakterielle Ursachen nachweisbar [12].
Seltener wird eine Parasiten- (Leishmaniose, Toxoplasmose) oder Pilzinfektion (Histoplasmose) als Ursache festgestellt. Trotzdem sollten Patienten mit Immundepression sowie immunkompetente Patienten, die sich vor Kurzem im Ausland aufhielten, systematisch darauf untersucht werden.
Neoplasien
MAS können sich bei Patienten entwickeln, die an einer Neoplasie leiden (insbesondere Hämopathien, seltener solide Tumoren). Aggressive Non-Hodgkin-Lymphome (vor allem T- oder NK-Zell-Lymphome) sind häufig als Ursache nachweisbar [13] und müssen im Falle eines MAS mit unklarer Ätiologie aktiv gesucht werden. Im Rahmen einer Krebserkrankung tritt das MAS im Allgemeinen in einem fortgeschrittenen oder rasch fortschreitenden Stadium auf.
Autoimmunerkrankungen
Ein MAS kann mit einer Autoimmunerkrankung assoziiert sein, am häufigsten mit dem systemischen Lupus erythematodes, dem Still-Syndrom (bzw. der entsprechenden Krankheit beim Kind, der juvenilen idiopathischen Arthritis) und der Kawasaki-Krankheit [16].
Andere
Es wurden Fälle von MAS beschrieben, die mit der Einnahme von Medikamenten (Antibiotika, Antiepileptika), einem schwerwiegenden chirurgischen Eingriff, schweren Verbrennungen [12] oder einer Transfusion assoziiert waren. In 15 bis 20% der Fälle ist keine Ursache feststellbar [16].
Klinisches Bild
Die Symptome des MAS sind unspezifisch und treten im Allgemeinen akut oder subakut auf. Bei der klinischen Untersuchung werden in den meisten Fälle ein anhaltendes Fieber (>38,5 °C) [12], eine ausgeprägte Asthenie und eine Hepatosplenomegalie festgestellt. In einem Drittel der Fälle werden periphere Adenopathien und neurologische Störungen beobachtet [20] (fokale Defizite, Enzephalopathien, Bewusstseinsstörungen bis hin zum Koma). Seltener wurden folgende Störungen beschrieben: dermatologische [13] (makulopapulöser Hautausschlag, Petechie, Purpura), pulmonale (interstitielle Infiltrate, die sich bis zu einem akuten Lungenödem entwickeln können [17, 18]) und digestive Symptome (Abdomenschmerzen, Durchfall [17]).
Labordiagnostik
Die Störung der hämatopoetischen Zelllinien ist ein Schlüsselelement der Diagnosestellung. Eine hypo- oder aregenerative, normochrome, normozytäre Anämie mit gestörten Hämolyseparametern (doch meist negativem Coombs-Test) ist konstant nachweisbar und geht in 80% der Fälle mit einer Thrombozytopenie einher [15]. Eine Panzytopenie kann ebenfalls auftreten. Gerinnungsstörungen sind möglich: Bei 50% der erwachsenen Patienten kommt es zu einer Hypofibrinogenämie und einem Anstieg der D-Dimer-Werte. Infolgedessen kann sich eine disseminierte intravasale Koagulopathie (DIC) entwickeln. In 60 bis 90% der Fälle wird eine Lebererkrankung mit unterschiedlicher Intensität beobachtet, die zuerst mit einer Zytolyse und in einer späteren Phase mit einer Cholestase verbunden ist und sich bis zu einer hepatozellulären Insuffizienz entwickeln kann [19, 20]. In signifikantem Ausmass nachweisbar sind überdies erhöhte LDH-Werte sowie eine sekundäre Hyponatriämie infolge einer inadäquaten ADH-Sekretion [20]. In zwei Drittel der Fälle tritt eine Hypertriglyzeridämie ohne Hypercholesterinämie auf, deren Ursache in der Hemmung der Lipoproteinlipase zu finden ist [12, 21]. Die ausgeprägte Hyperferritinämie kann dabei helfen, das MAS von anderen systemischen Entzündungssyndromen zu unterscheiden. Sie wird dadurch verursacht, dass Ferritin durch die Makrophagen und Hepatozyten sowie bei der Phagozytose der Blutzellen freigesetzt wird [22]. Laut einer Studie weist eine Ferritinämie von über 10000 µg/l in der pädiatrischen Population mit 90%iger Sensitivität und 96%iger Spezifität auf ein MAS hin [23]. Eine andere Studie zeigte einen signifikanten Unterschied zwischen den Ferritin-Werten von MAS-Patienten und jenen der Kontrollgruppe [24]. Beim Verdacht auf ein MAS spricht – bei gleichzeitigem Fehlen einer ausgeprägten Hyperferritinämie (>5000 µg/l) – eine verringerte Konzentration des glykosylierten Ferritins (<20%) für die Bestätigung der Diagnose [25]. Eine hohe Spezifität weisen folgende Spezialbefunde auf [27]: Erhöhung der Plasmakonzentration der löslichen α-Kette des Interleukin-2-Rezeptors (sCD25), Erhöhung der CD163-Serumkonzentration [26] und Verringerung der NK-Zell-Funktion.
Histologie und Zytologie
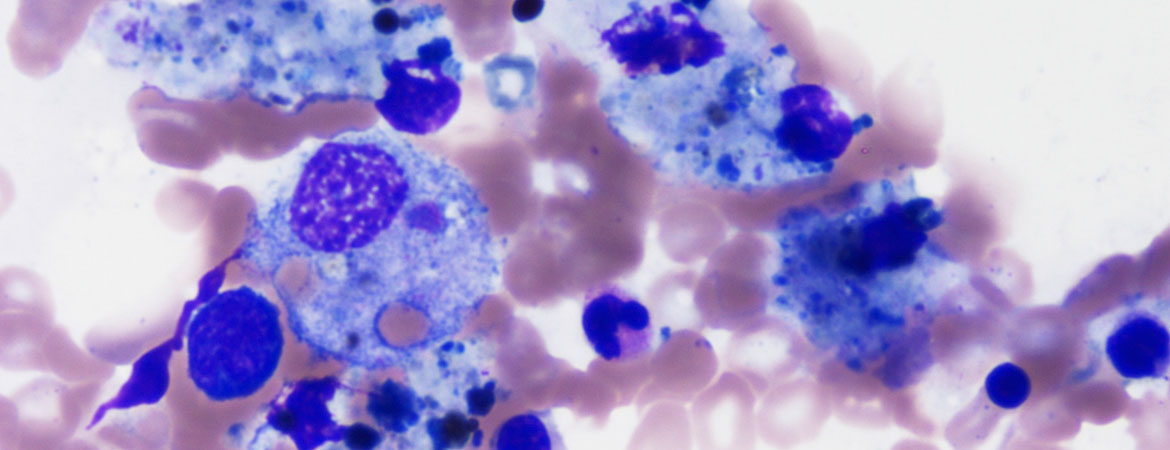
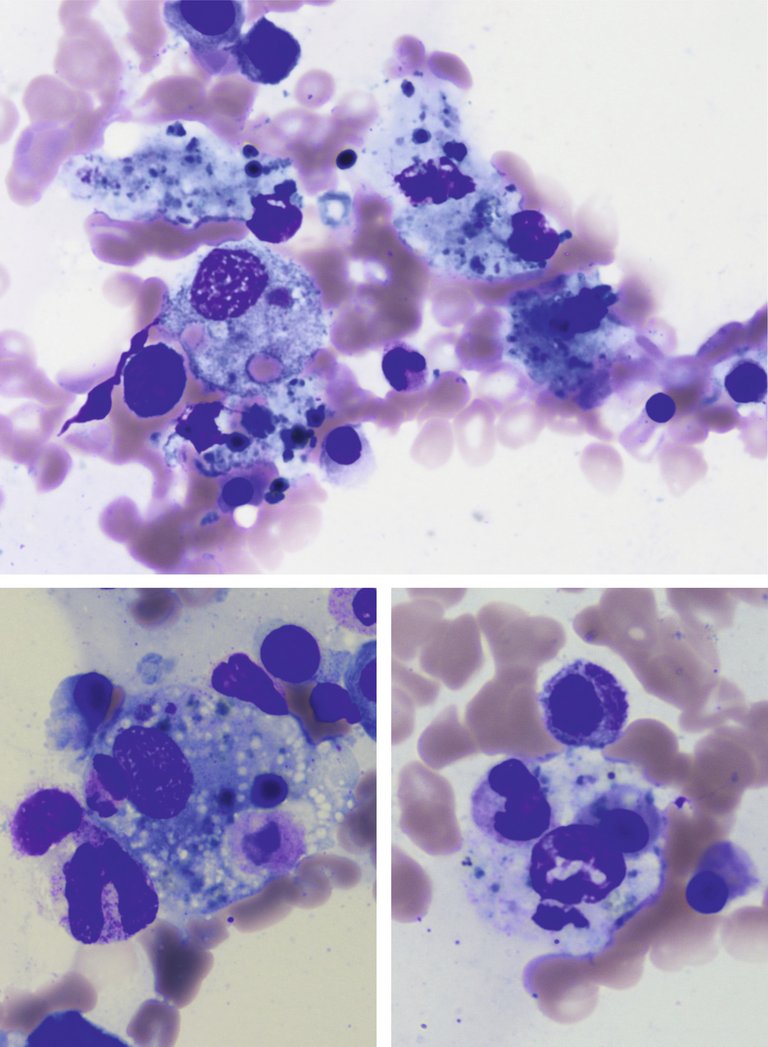
Bei der zytologischen Untersuchung ist in der Mehrheit der Fälle ein zellarmes Knochenmark mit einer relativen Vermehrung der Histiozyten festzustellen. Das zytologische Erscheinungsbild ist gutartig und weist auf eine Hämophagozytose hin: Die intrazytoplasmatischen Vakuolen der Histiozyten enthalten Blutzellen und ihre Vorläufer (Erythrozyten, Erythroblasten, Granulozyten, Thrombozyten, Lymphozyten) (Abb. 1). Die Hämophagozytose ist ein wichtiges Element zur Diagnose eines MAS, muss allerdings im klinischen Kontext betrachtet werden: Sie ist im Fall einer Bluttransfusion, einer Autoimmunerkrankung oder einer Sepsis zu finden [28]. In 45% der Fälle kann sie zu Beginn der Symptome fehlen, während sie bei späteren Untersuchungen nachweisbar ist [11].
Abbildung 1: Hämatomyelogramm mit May-Grünwald-Giemsa-Färbung (Vergrösserung 500-fach). Darstellung einer Hämophagozytose: Makrophagen phagozytieren Blutzellen (veröffentlicht mit freundlicher Genehmigung von Dr. med. Kaveh Samii, Service d’hématologie, HUG, Genf, Schweiz).
Die zytologische Untersuchung erfolgt üblicherweise in Form eines Hämatomyelogramms, das eine höhere Sensitivität als die osteomedulläre Biopsie aufweist, um die intramedulläre Hämophagozytose nachzuweisen [16]. Der Grenzwert eines 2- bis 3%igen Anteils aktivierter Makrophagen (mit dem Erscheinungsbild einer Hämophagozytose) wird oftmals als Diagnosekriterium herangezogen.
Weitere zytologische Untersuchungen (ganglionäre Zytopunktion, Leberbiopsie, Liquor cerebrospinalis) können zusätzliche Anhaltspunkte liefern.
Diagnose
1991 schlug die «Histiocyte Society» Diagnosekriterien vor, die 2004 aktualisiert wurden (Tab. 2): Mit hoher Wahrscheinlichkeit liegt ein MAS vor, wenn fünf von acht Kriterien erfüllt sind. Die Kriterien galten zuerst für die pädiatrische Population, werden aber oftmals für erwachsene Patienten extrapoliert. Sie umfassen Analysen (wie die Untersuchung der zytotoxischen Aktivität der NK-Zellen), die bei den sekundären Formen der Krankheit wenig sinnvoll sind, in nicht spezialisierten Kliniken selten zur Verfügung stehen und in der Routinepraxis kaum durchgeführt werden. Nach Bewertung (mittels Delphi-Studie) der Elemente, auf die sich eine Gruppe von Experten in der klinischen Praxis stützt, um beim Erwachsenen ein MAS zu diagnostizieren [46], haben Fardet und Mitarbeiter einen neuen Diagnosescore vorgeschlagen und validiert: den HScore [29] (Tab. 2, unterer Abschnitt, http://saintantoine.aphp.fr/score/). Der Score basiert auf allgemein verfügbaren klinischen und labormedizinischen Elementen, bezieht erstmals eine bekannte Immundepression in die Kriterien ein und unterstreicht dadurch die Bedeutung prädisponierender ätiologischer Faktoren.
Tabelle 2: Kriterien zur Diagnose des Makrophagenaktivierungssyndroms.
Hämophagozytose laut Hämatomyelogramm (nein [0 Punkte], ja [35 Punkte])
Die Wahrscheinlichkeit eines MAS variiert zwischen <1% (bei einem HScore von <90) und>99% (bei einem HScore von >250) Maximaler HScore: 337 Punkte
Behandlung
Unbehandelt verläuft ein MAS meist innert einiger Monate tödlich [30], weshalb aggressive Diagnose- und Therapiemassnahmen gerechtfertigt sind. Die Therapie beruht auf drei Grundlagen: Behandlung der Symptome, Ausschaltung der Ursache sowie Unterdrückung der T-Zell-Aktivierung und der Entzündungsreaktion.
Die Symptombehandlung umfasst je nach Situation: Transfusionen, Ausgleich der Gerinnungsstörung sowie des Wasser- und Elektrolythaushalts, Breitbandantibiotika (im Falle einer febrilen Neutropenie) [31].
Wenn als Ätiologie eine Infektion oder eine onkologische Erkrankung nachweisbar ist, muss die therapeutische Bekämpfung der Ursache rasch begonnen werden [32].
Für die Behandlung des primären MAS wurde ein spezielles Protokoll namens HLH-2004 entwickelt [6]: Es beruht auf der Kombination von Dexamethason, Etoposid, Ciclosporin und Methotrexat (intrathekal), gefolgt von einer allogenen Knochenmarktransplantation. Bei einem sekundären MAS wurde dieses Protokoll nicht validiert.
Über die Behandlung der sekundären Form steht keine randomisierte Prospektivstudie zur Verfügung [12].
– Etoposid (VP-16) ist aufgrund der selektiven Wirkung auf aktivierte CD8-T-Lymphozyten [33] ein Mittel erster Wahl zur Behandlung des MAS [32]. Es gehört zur Klasse der Topoisomerase-II-Hemmer. Etoposid ist rasch wirksam (üblicherweise innert 48 Stunden), gut verträglich und bewirkt als einzige Therapie eine erwiesene Verringerung der Mortalität: Laut Imashuku et al. [34] steht die Anwendung von Etoposid bei krebs- oder infektionsbedingtem (besonders EBV-bedingtem) MAS im Zusammenhang mit einer Überlebensrate von 90% (gegenüber 56%, wenn es nicht angewandt wird). Die allfälligen Nachteile (Risiko hämatologischer Toxizität, Notwendigkeit der Dosisanpassung im Falle einer Nierenfunktionsstörung) werden durch den Nutzen aufgewogen (Verringerung der Organstörungen, Verbesserung der Entzündungsparameter) [35].
– Bei autoimmun [36] und autoinflammatorisch bedingtem sowie bei lymphomassoziiertem MAS [37] wird im Allgemeinen eine intravenöse, hochdosierte Kortikoidtherapie eingesetzt, gleichwohl wurde dafür noch kein Nutzen im Hinblick auf die Mortalität nachgewiesen [35]. Bei diesen Indikationen kann sie mit Etoposid kombiniert werden [38]. Im Falle einer Störung des Zentralnervensystems wird ebenfalls die Anwendung von Dexamethason empfohlen, da es die Blut-Hirn-Schranke überwinden kann [39]. Abgesehen von diesen Situationen ist die Indikation der Kortikoidtherapie aufgrund des Infektionsrisikos weniger eindeutig [37].
– Ciclosporin ist ein starker Inhibitor der Proliferation der T-Lymphozyten und ist von Interesse, falls die Etoposid-Therapie versagt [35] oder eine Autoimmunerkrankung festgestellt wird [40]: Bei autoimmun bedingtem MAS steht es im Zusammenhang mit einer Überlebensrate von 76% [12]. Der Hauptnachteil ist das Risiko neurologischer und renaler Toxizität.
– Bei refraktärem MAS sowie in Sonderfällen werden monoklonale Antikörper eingesetzt, allerdings ist noch kein Nutzen im Hinblick auf die Überlebensrate belegt: Antikörper gegen Interleukin-1r und Interleukin-6 bei MAS, das mit dem Still-Syndrom assoziiert ist; Rituximab bei MAS, das durch ein diffuses grosszelliges B-Zell-Lymphom, systemischen Lupus erythematodes oder EBV ausgelöst wurde und gegenüber der oben genannten Behandlung refraktär ist [12]. Die gezielte Behandlung primärer MAS-Formen beim Kind mit Anti-IFNγ-Antikörpern (NI-0501) ist derzeit Gegenstand einer klinischen Studie (NCT01818492) und wird in naher Zukunft auch bei sekundärem, neoplasiebedingtem MAS untersucht. Darüber hinaus werden zurzeit Therapien, die auf den TLR-9 und den MyD88-abhängigen Signalweg (siehe oben) abzielen, in tierexperimentellen Studien getestet [9, 10, 11].
– Die Gabe intravenöser Immunglobuline wurde vorgeschlagen [41]. Diesbezüglich stehen jedoch nur sehr wenige Daten zur Verfügung. Sie können infrage kommen, wenn eine virale Erstinfektion oder ein Still-Syndrom festgestellt wurden und keine schwerwiegenden Symptome vorliegen [11].
Die Indikationen sind in Tabelle 3 zusammengefasst.
Tabelle 3: Spezifische Therapien gegen das sekundäre Makrophagenaktivierungssyndrom (MAS) beim Erwachsenen.
Therapie
Indikationen
Etoposid (VP-16)
Mittel erster Wahl – EBV-bedingtes MAS – MAS mit schwerwiegenden klinischen und laborchemischen Befunden
Hochdosierte Kortikoidtherapie
Autoimmun oder autoinflammatorisch bedingtes MAS
Lymphomassoziiertes MAS
Dexamethason bei Störung des Zentralnervensystems
Ciclosporin
Durch eine Autoimmunerkrankung ausgelöstes MAS
Nach Versagen der Etoposid-Therapie (strittige Indikation)
Rituximab (Anti-CD20-Antikörper)
Durch systemischen Lupus erythematodes ausgelöstes MAS
Durch EBV-Infektion ausgelöstes MAS
Durch diffuses grosszelliges B-Zell-Lymphom ausgelöstes MAS
Antikörper gegen Interleukin-1r und Interleukin-6
Mit dem Still-Syndrom assoziiertes MAS
Intravenöse Immunglobuline
Sekundäres MAS infolge einer Virusinfektion (wenige Daten über den Nutzen)
Anti-IFNγ-Antikörper
Laufende Studien
Abkürzung: EBV = Epstein-Barr-Virus
Verlauf und Prognose
Die Krankheit kann sich unterschiedlich entwickeln: Bei einem positiven Verlauf klingen die Symptome und labormedizinischen Störungen in einem Zeitraum von 7 bis 56 Tagen ab [19]. Nach der Akutphase hängt die Mortalität von den Nebenwirkungen der Immunsuppression und dem Auftreten opportunistischer Infektionen ab. Es ist möglich, dass ein sekundäres MAS rezidiviert, nachdem es anfangs gut ansprach.
Die Prognose ist von einer hohen Letalität gekennzeichnet und bei Erwachsenen beträgt die Mortalität 40% [12]. Sie hängt jedoch von der Grunderkrankung ab: Bei sekundärem MAS im Zusammenhang mit einer Autoimmunerkrankung oder einer Virusinfektion ist die Prognose besser als bei neoplasieassozierten Formen [42]. Zu den Faktoren, die mit grösserer Mortalität verbunden sind, zählen: höheres Alter (>50 Jahre bei der Diagnose [43]), Bestehen von Zytopenien, Hyperferritinämie (>50 000 µg/l [43]), Auftreten einer disseminierten intravasalen Koagulopathie, intrahepatische Cholestase, erhöhter Interferon-γ-Wert [44]. Wenn nach Beginn der Behandlung das Fieber, die Anämie und die Thrombozytopenie nicht abklingen, deutet dies ebenfalls auf eine negative Prognose hin [45].
Fazit
Das reaktive MAS ist nach wie vor eine unterdiagnostizierte Krankheit, die unbehandelt einen tödlichen Verlauf nehmen kann. Die Therapie muss möglichst rasch begonnen werden, in manchen Fällen sogar vor Feststellung der Ursache. Im Hinblick auf die Festlegung zuverlässiger Diagnosekriterien und standardisierter Behandlungsprotokolle müssen noch Fortschritte erzielt werden. Im Mittelpunkt der aktuellen Forschung über das MAS stehen die pathophysiologischen Mechanismen, die Erfassung von Genmutationen und die prädisponierenden Faktoren.
Das Wichtigste für die Praxis
• Das Makrophagenaktivierungssyndrom (MAS) ist eine schwere, oft nicht richtig erkannte Krankheit mit vielfältigen und unspezifischen Symptomen, die tödlich verlaufen kann. Das MAS kann primär sein oder sekundär infolge anderer Krankheiten auftreten.
• Das MAS wird durch die mangelhafte Zytotoxizität der T-Lymphozyten und/oder NK-Zellen ausgelöst, wodurch es zu einer massiven Produktion proinflammatorischer Zytokine und Aktivierung der Makrophagen kommt. Die Deregulierung der Toll-like-Rezeptoren und des MyD88-abhängigen Signalwegs scheint ebenfalls die Entstehung des Syndroms zu beeinflussen.
• Eine ausgeprägte Hyperferritinämie (>5000 µg/l) und eine Hypertriglyzeridämie sind Symptome, die im Zusammenhang mit weniger spezifischen Störungen (Bi- oder Panzytopenie, gestörte Leberfunktion, Gerinnungsstörung, erhöhte LDH-Werte) auf diese Diagnose hindeuten.
• Ein zentrales Element der Diagnose ist der Nachweis einer Hämophagozytose aufgrund histologischer oder zytologischer Untersuchungen. Scores (HLH-2004-Kriterien, HScore) dienen als Diagnoseinstrumente.
• Die sekundären Formen des MAS treten reaktiv infolge von Infektionen, Neoplasien oder Autoimmunerkrankungen auf.
• In Ermangelung randomisierter Prospektivstudien ist die Therapie des MAS noch ungenügend belegt. Sie muss aggressiv sein und beruht auf der Behandlung der Symptome, der Ausschaltung der Ursache und der Unterdrückung der T-Zell-Aktivierung und der Entzündungsreaktion. Als spezifische Therapie werden Etoposid (VP-16), Kortikoide, Ciclosporin und monoklonale Antikörper vorgeschlagen, weitere Arzneistoffe mit gezielter Wirkung sind derzeit Gegenstand von Studien.
• Die Prognose hängt von der auslösenden Grunderkrankung ab, die durchschnittliche Mortalität beim Erwachsenen beträgt 40%.
Disclosure Statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Korrespondenz
Dr. med Henri Lu Médecin-assistant Service de Médecine interne CHUV, Lausanne Rue du Bugnon 46 CH-1011 Lausanne henri.lu[at]me.com
Empfohlene Literatur
– Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31.
– Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. The Lancet. 2014;383(9927):1503–16.
– Rivière S, Galicier L, Coppo P, Marzac C, Aumont C, Lambotte O, et al. Reactive Hemophagocytic Syndrome in Adults: A Retrospective Analysis of 162 Patients. Am J Med. 2014;127(11):1118–25.
– Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and Validation of the HScore, a Score for the Diagnosis of Reactive Hemophagocytic Syndrome: Score for Reactive Hemophagocytic Syndrome. Arthritis Rheumatol. 2014;66(9):2613–20.
1 Scott RB, Robb-Smith AH. Histiocytic medullary reticulocytosis. Lancet. 1939;2:194–8.
2 Karras A, Thaunat O, Noël LH, Delahousse M. Syndrome d’activation macrophagique: implications pour le néphrologue. Actualités néphrologiques. 2005;59–80.
3 Meki A, O’Connor D, Roberts C, et al. Hemophagocytic lymphohistiocytosis in chronic lymphocytic leukemia. J Clin Oncol. 2011;29(24):e685–7.
4 Machaczka M, Vaktnäs J, Klimkowska M, Hägglund H Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52:613–9.
5 Kelesidis T, Humphries R, Terashita D, et al. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in Los Angeles County. J Med Virol. 2012;84:777–85.
6 Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31.
7 Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood. 2011;118:618–26.
8 Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. British Journal of Haematology. 2013 Jun;161(5):609–22.
9 Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, et al. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. Journal of Clinical Investigation. 2011 Jun 1;121(6):2264–77.
10 Krebs P, Crozat K, Popkin D, et al. Disruption of MyD88 signaling suppresses hemophagocytic lymphohistiocytosis in mice. Blood. 2011;117:6582–8.
11 Canna SW, et al. Interferon-gamma mediates anemia but is dispensable for fulminant toll-like receptor 9-induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum. 2013 Jul;65(7):1764–75.
12 Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. The Lancet. 2014;383(9927):1503–16.
13 Campo M, Berliner N. Hemophagocytic Lymphohistiocytosis in Adults. Hematology/Oncology Clinics of North America. 2015 Oct;29(5):915–25.
14 Tseng YT, Sheng WH, Lin BH, et al. Causes, clinical symptoms, and outcomes of infectious diseases associated with hemophagocytic lymphohistiocytosis in Taiwanese adults. J Microbiol Immunol Infect. 2011;44:191–7.
15 Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. The Lancet infectious diseases. 2007;7(12):814–22.
16 Lambotte O, Méchaï F. Syndrome d’activation macrophagique. La Lettre de l’infectiologue [Internet]. 2007;22(3).
17 Karras A, Thervet E, Legendre C, and the Groupe Coopératif de transplantation d’Ile de France. Hemophagocytic syndrome in renal transplant recipients: report of 17 cases and review of literature. Transplantation. 2004;77:238–43.
18 Majluf-Cruz A, Sosa-Camas R, P.rez-Ram.rez O, Rosas-Cabral A, Vargas-Vorackova F, Labardini-M.ndez J. Hemophagocytic syndrome associated with hematological neoplasias. Leuk Res. 1998;22:893–8.
19 Papo T. Syndromes hémophagocytaires, syndrome d’activation des macrophages. EMC-AKOS (Traité de médecine). 2011;1–8. SAM 2.
20 Rivière S, Galicier L, Coppo P, Marzac C, Aumont C, Lambotte O, et al. Reactive Hemophagocytic Syndrome in Adults: A Retrospective Analysis of 162 Patients. The American Journal of Medicine. 2014 Nov;127(11):1118–25.
21 Okamoto M, Yamaguchi H, Isobe Y, Yokose N, Mizuki T, Tajika K, et al. Analysis of triglyceride value in the diagnosis and treatment response of secondary hemophagocytic syndrome. Internal Medicine. 2009;48(10):775–81.
22 Knovich MA, Storey JA, Coff man LG, Torti SV, Torti FM. Ferritin for the clinician. Blood Rev. 2009;23:95–104.
23 Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50:1227–35.
24 Francois B, Trimoreau F, Vignon P, et al. Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating factor. Am J Med. 1997;103:114–20.
25 Wang Z, Wang Y, Wang J, et al. Early diagnostic value of low percentage of glycosylated ferritin in secondary hemophagocytic lymphohistiocytosis. Int J Hematol. 2009;90:501–5.
26 Schaer DJ, Schleiffenbaum B, Kurrer M, et al. Soluble hemoglobin-haptoglobin scavenger receptor CD163 as a lineage-specific marker in the reactive hemophagocytic syndrome. Eur J Haematol. 2005;74(1):6–10.
27 Imashuku S, Teramura T, Tauchi H, IshidaY, OtohY, Sawada M, et al. Longitudinal follow-up of patients with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Haematologica. 2004;89:183–8.
28 Jordan MB, Allen CE, Weitzman S, et al. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041–52.
29 Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and Validation of the HScore, a Score for the Diagnosis of Reactive Hemophagocytic Syndrome: Score for Reactive Hemophagocytic Syndrome. Arthritis & Rheumatology. 2014 Sep;66(9):2613–20.
30 Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine 2014;93:100–5.
31 Buyse S, Teixeira L, Galicier L, et al. Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Med. 2010;36:1695–702.
32 Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood. 2015;125(19):2908–14.
33 Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, Jordan MB. Etoposide Selectively Ablates Activated T Cells To Control the Immunoregulatory Disorder Hemophagocytic Lymphohistiocytosis. The Journal of Immunology. 2014 Jan 1;192(1):84–91.
34 Imashuku S, Kuriyama K, Teramura T, et al. Requirement for etoposide in the treatment of Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis. J Clin Oncol 2001;19:2665–73.
35 Galicier L, Ce qu’il faut savoir sur le syndrome d’activation macrophagique aux soins intensifs. Réanimation. 2014;23:482–90.
36 Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology (Oxford). 2008;47:1686–91.
37 Larroche C. Hemophagocytic lymphohistiocytosis in adults: Diagnosis and treatment. Joint Bone Spine. 2012 Jul;79(4):356–61.
38 Tothova Z, Berliner N. Hemophagocytic syndrome and critical illness: new insights into diagnosis and management [published online ahead of print January 8, 2014]. J Intensive Care Med. doi:10.1177/0885066613517076.
39 Henter JI, Tondini C, Pritchard J. Histiocyte disorders. Crit Rev Oncol Hematol. 2004;50:157–74.
40 Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002;14:548–52.
41 Larroche C, Bruneel F, André MH, et al (2000). [Intravenously administered gamma-globulins in reactive hemaphagocytic syndrome. Multicenter study to assess their importance, by the immunoglobulins group of experts of CEDIT of the AP-HP]. Ann MedInterne (Paris) 151:533–9.
42 Takahashi N, Chubachi A, KumeM, et al. A clinical analysis of 52 adult patients with hemophagocytic syndrome: the prognostic significance of the underlying diseases. Int J Hematol. 2001;74:209–13.
43 Tseng YT, Sheng WH, Lin BH, et al. Causes, clinical symptoms, and outcomes of infectious diseases associated with hemophagocytic lymphohistiocytosis in Taiwanese adults. J Microbiol Immunol Infect. 2011;44(3):191–7.
44 Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2015;90:220–4.
45 Trottestam H, Berglöf E, Horne A, et al. Risk factors for early death in children with haemophagocytic lymphohistiocytosis. Acta Paediatr. 2012;101(3):313–8.
46 Hejblum G, Lambotte O, Galicier L, Coppo P, Marzac C, Aumont C, Fardet L. A web-based delphi study for eliciting helpful criteria in the positive diagnosis of hemophagocytic syndrome in adult patients. PLoS One. 2014;9(4):e94024.