a Service de médecine interne, Centre Hospitalier Universitaire Vaudois (CHUV); b Service de pneumologie, Centre Hospitalier Universitaire Vaudois (CHUV); c Servizio de pneumologia, Ospedale Regionale di Mendrisio
Ein 18-jähriger Landschaftsgärtner kommt aufgrund seit einigen Stunden bestehender moderater Hämoptysen in die Notaufnahme eines Regionalspitals. Er leidet trotz empirischer Antibiotikabehandlung seit sechs Wochen an zunehmender Asthenie und Dyspnoe. Der junge Mann weist einen guten Gesundheitszustand auf, mit Ausnahme eines Asthmas, welches er bei Bedarf mit inhalativen Kortikoiden und Beta-2-Mimetika behandelt. Andere Medikamente nimmt er nicht ein. Er raucht Tabak und gelegentlich Marihuana. Der Patient hat weder eine Reise unternommen, noch war er inhaftiert. Er gibt an, dass er beruflich gegenüber Pestiziden exponiert sei, jedoch eine entsprechende Atemschutzmaske verwende.
Die klinische Untersuchung ist unauffällig mit Vitalparametern im Normbereich. Die Blutuntersuchung ergibt ein leicht erhöhtes Gesamtbilirubin und eine regenerative mikrozytäre hypochrome (Retikulozyten 28/1000, bei einem Normwert von 5–15/1000), jedoch nicht hämolytische Anämie (LDH und Haptoglobin im Normbereich) als einzige Abweichungen des Routinelabors (Hämostase und Elektrolytwerte normal, Nierenfunktion erhalten, C-reaktives Protein mit 10 g/l im oberen Normbereich, HIV-Test negativ). Urinsediment und Spot-Urin sind unauffällig.
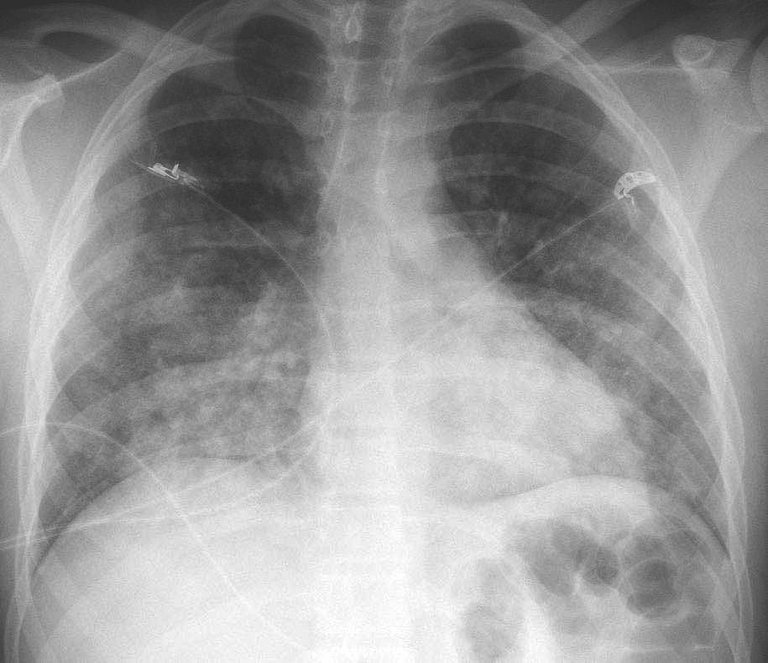
Frage 1: Die Thorax-Röntgenaufnahme zeigt beidseitige basale alveoläre Infiltrate (Abb. 1). Was ist Ihr erster diagnostischer Schritt?
Abbildung 1: Thorax-Röntgenaufnahme a.p. des Patienten.
a) Eine Bronchoskopie
b) Eine Thorax-Computertomographie mit Kontrastmittel
c) Eine empirische Antibiotikatherapie zu Diagnosezwecken
d) Ein T-Spot.TB-Test und eine Aerosolisolation
e) Eine Konsultation eines Hals-Nasen-Ohrenarztes (HNO)
Hämoptysen sind ein medizinischer Notfall mit einer hohen Morbidität und Mortalität, der umso dringender behandelt werden muss, je stärker der Blutverlust ist. Die Manifestationen können mehr oder weniger ausgeprägt sein und sich manchmal in Form von Ersticken oder hämorrhagischem Schock äussern. In diesem Fall sind eine orotracheale Intubation zum Schutz der nicht betroffenen Atemwege, eine Bronchoskopie oder radiologisch-interventionelle Massnahmen erforderlich, um die Blutung zu stoppen. Bei geringerer Ausprägung (Dyspnoe, Anämie oder isolierte Veränderungen im Röntgenbild) sollte zuallererst eine Ursache im HNO- oder gastroenterologischen Bereich ausgeschlossen werden, bevor weitere Untersuchungen der Lunge erfolgen.
In 80–90% der Fälle kann die Blutungsquelle durch eine Computertomographie (CT) des Thorax mit Kontrastmittel lokalisiert werden. Bei unserem Patienten zeigt dieses beidseitige Infiltrate und führt zum Ausschluss einer Lungenembolie (Abb. 2).
Wenn die Blutungsquelle durch die vorhergehenden Untersuchungen nicht identifiziert werden konnte, ist eine Bronchoskopie indiziert. Auf diese Weise kann durch Rot- oder Rosafärbung der bronchoalveolären Lavage (BAL) bestätigt werden, dass sich die Blutungsquelle in der Lunge befindet, eine bronchiale Blutung lokalisiert und behandelt sowie nach einer infektiösen oder immunbedingten Ursache gesucht werden.
Obgleich eine Bronchitis in dieser Altersklasse die häufigste Ursache für Hämoptysen darstellt, ist erstere üblicherweise viral bedingt und erfordert keine empirische Antibiotikatherapie. Gelegentlich tritt eine bakterielle Superinfektion als Komplikation auf.
Eine Lungentuberkulose ist bei einem jungen, immunkompetenten Schweizer extrem unwahrscheinlich. Der T-Spot.TB-Test, der HIV-Test und die Untersuchung auf säure- und alkoholfeste Stäbchen sind negativ.
Aufgrund dessen ist zunächst eine HNO-Untersuchung durchzuführen. Diese erweist sich als unauffällig, ermöglicht jedoch den Ausschluss einer posterioren Epistaxis oder einer Blutung im Hals-Rachen-Bereich, welche bei Aspiration in der Röntgenaufnahme wie eine alveoläre Hämorrhagie (AH) aussehen können. HNO-Ereignisse in der Vorgeschichte oder der Geschmack von Blut im Mund können auf eine derartige Ursache hinweisen. Das Erbrechen von Blut (Hämatemesis) kann mitunter schwer vom Bluthusten (Hämoptyse) zu unterscheiden sein.
Frage 2: Die Bronchoskopie bestätigt eine alveoläre Hämorrhagie. Welches ist die wahrscheinlichste Ursache?
a) Eine Toxinexposition
b) Eine Leptospirose
c) Eine Vaskulitis
d) Ein Bronchialkarzinom
e) Eine angeborene Mitralklappenstenose
Die detaillierte Anamnese ergibt eine berufliche Exposition gegenüber Pestiziden. Die Verwendung einer entsprechenden Atemschutzmaske und die Exposition im Freien sprechen jedoch eher gegen diese Hypothese. Sechs Wochen vor den Hämoptysen hat der Patient eine grössere Menge Marihuana durch das Einatmen kalten Rauchs aus einer «Bong» konsumiert. Obgleich in diesem Zusammenhang einige AH-Fälle beschrieben werden, treten sie für gewöhnlich sehr rasch nach dem Konsum auf. Zudem hat der Patient nie Heroin konsumiert, was eine weitere mögliche AH-Ursache sein kann.
Aufgrund seines Berufs, bei dem ein Kontakt mit Nagerurin möglich ist, könnte eine ikterohämorrhagische Leptospiroseform (Weil-Krankheit) vermutet werden. Diese hat jedoch ausgeprägtere klinische Manifestationen zur Folge und beeinträchtigt typischerweise die Nieren- und Leberfunktion. Die serologische Untersuchung hat während der septikämischen Phase lediglich eine Sensitivität von 30% (63% während der Immun- und 76% während der Rekonvaleszenzphase) und wird nach und nach durch PCR-Tests ersetzt [2]. Eine weitere infektiöse Ursache wie Legionnellapneumophila kann durch einen negativen Antigennachweis im Urin und eine sterile BAL zuverlässig ausgeschlossen werden.
Nicht selten (in 19–42%) ist eine Autoimmunkrankheit die Ursache einer AH. Mikroskopische Polyangiitis, Granulomatose mit Polyangiitis (Wegener-Granulomatose), systemischer Lupus erythematodes, das Goodpasture-Syndrom und seltener andere Kollagenosen (rheumatoide Polyarthritis, Sklerodermie, entzündliche Myopathien und Mischkollagenosen) oder Vaskulitiden (eosinophile Granulomatose mit Polyangiitis, früher Churg-Strauss-Syndrom genannt, Purpura Schönlein-Henoch und IgA-Nephropathie) sind diagnostisch abzuklären.
Ein Bronchialkarzinom oder eine andere pulmonale Neoplasie erscheinen angesichts der medizinischen Vorgeschichte ohne weitere Alarmzeichen, des jungen Alters des Patienten und einer Blutung, die hauptsächlich im Bronchialbaum lokalisiert sein dürfte, am unwahrscheinlichsten. Auch die Thorax-CT und die Bronchoskopie liefern diesbezüglich keine weiteren Anhaltspunkte.
Herzkrankheiten sind, neben Lungeninfektionen, Drogen- oder Toxinexposition sowie Störungen des ex- oder intrinsischen Gerinnungssystems (Antiphospholipid-Syndrom, thrombotisch-thrombozytopenische Purpura), die häufigste nicht immunbedingte Ursache für AH. Mittels Echokardiographie wird eine angeborene Mitralklappenstenose ausgeschlossen, die für eine Herzinsuffizienz mit postkapillärer pulmonaler Hypertonie verantwortlich sein könnte. Eine bronchovaskuläre Fistel, eine Hämosiderose oder eine pulmonale Endometriose kommen seltener vor.
Frage 3: Somit bleibt noch die Hypothese einer Vaskulitis bestehen. Welcher Test ist in diesem Untersuchungsstadium am sinnvollsten?
a) Ein indirekter Immunfluoreszenztest (IFT) auf antinukleäre Antikörper (ANA)
b) Ein IFT auf antizytoplasmatische Antikörper der neutrophilen Granulozyten (ANCA)
c) Ein Test auf glomeruläre Basalmembran-Antikörper (Anti-GBM)
d) ELISA-Tests auf Antikörper gegen Myeloperoxidase (MPO) und Proteinase 3 (PR3)
e) Bestimmung der Eosinophilen im Blutplasma und des Gesamt-IgE
Ein Test auf ANA (Abb. 3) ist bei febriler Arthritis, palpabler Purpura, Sicca-Syndrom, Morbus Raynaud, (immunbedingter) Zytopenie oder Glomerulonephritis sinnvoller als bei einer isolierten AH. Er dient vor allem zur Diagnostik von Kollagenosen wie systemischem Lupus erythematodes (SLE), Sjögren-Syndrom oder Sklerodermie mit einer Sensitivität und Spezifizität von ca. 50% (mit Ausnahme von SLE mit einer Sensitivität von 95%). Bei einem negativen Resultat muss kein ELISA-Test («enzyme-linked immunosorbent assay») zur Differenzierung in Antikörper gegen doppelsträngige DNA (anti-dsDNA) und Nukleoproteinantikörper durchgeführt werden.
Abbildung 3: Vorgeschlagener Algorithmus für die Suche nach antinukleären Antikörpern (ANA) und antizytoplasmatischen Antikörpern der neutrophilen Granulozyten (ANCA) mittels direktem Immunfluoreszenztest (angepasst und vereinfacht gemäss [10]). PR3 = Proteinase 3; MPO = Myeloperoxidase.
Bei AH, pneumorenalem Syndrom und multiplen Lungenknötchen ist ein ANCA-Test prioritär. ANCA sind vor allem mit mikroskopischer Polyangiitis, Granulomatose mit Polyangiitis (Wegener-Granulomatose) oder eosinophiler Granulomatose mit Polyangiitis (EGPA = Churg-Strauss-Syndrom) assoziiert und werden gelegentlich beim Goodpasture-Syndrom in Kombination mit anti-GBM beobachtet. Bei unserem Patienten ist der indirekte Immunfluoreszenztest (IFT) auf ANCA negativ.
Der Test auf anti-GBM-Antikörper ist ein unerlässlicher Marker für die Diagnose des Goodpasture-Syndroms. Bei unserem Patienten wurde dieser zweimal durchgeführt und beide Male war das Ergebnis negativ.
Bei erhöhtem ANCA-Titer hat ein Test auf anti-MPO-Antikörper, insbesondere bei mikroskopischer Polyangiitis, eine wichtige diagnostische Bedeutung, während die anti-P3-Antikörper vor allem bei Granulomatose mit Polyangiitis und seltener bei mikroskopischer Polyangiitis und segmental nekrotisierender Glomerulonephritis positiv getestet werden.
Eine Eosinophilie im Blut und ein Anstieg des Gesamt-IgE sind in zahlreichen Situationen wie beispielsweisebei saisonalen allergischen Reaktionen zu beobachten und somit für den Nachweis einer Vaskulitis weniger aussagekräftig. Da weder eine Eosinophilie, noch eine Beteiligung der Nasennebenhöhlen oder eine Neuropathie vorliegen, ist eine EGPA, die aufgrund des Asthmas und der vorübergehenden Lungeninfiltrate vermutet werden könnte, weniger wahrscheinlich. In diesem Stadium erscheint die Möglichkeit eines Goodpasture-Syndroms mit isolierter Lungenbeteiligung aufgrund des schrittweisen Ausschlusses anderer Ursachen und eines mit jedem Test weiter ansteigenden, wenn auch noch normalen, anti-GBM-Antikörperspiegels am wahrscheinlichsten.
Frage 4: Welches ist der beste diagnostische Test zur Bestätigung eines Goodpasture-Syndroms bei unserem Patienten?
a) Der Test auf anti-GBM-Antikörper anhand einer Lungenbiopsie
b) Die Quantifizierung der anti-GBM-Antikörper im Blut mittels ELISA-Test
c) Der Nachweis von anti-GBM-Antikörpern im Blut mittels direktem Immunfluoreszenztest
a) Der Test auf anti-GBM-Antikörper anhand einer Nierenbiopsie
e) Der Test auf durch Kollagenase solubilisierte GBM mittels Western Blot
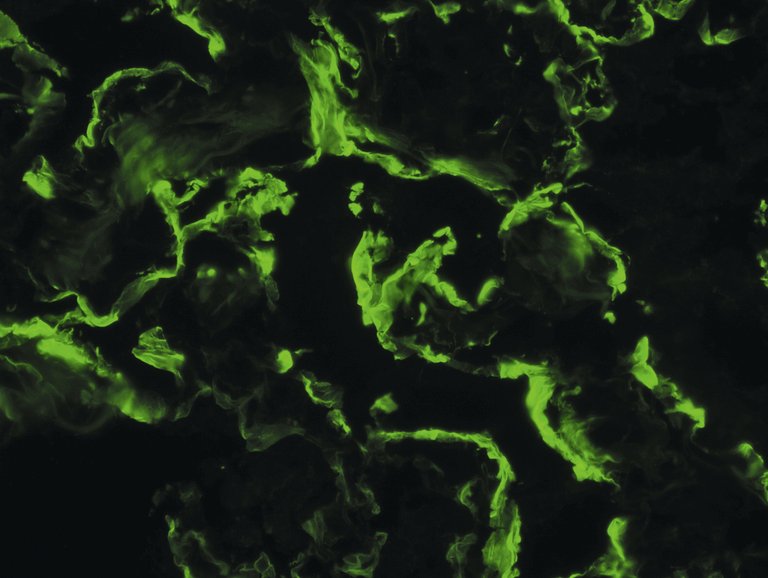
Die Biopsie des betroffenen Organs ist der diagnostische Goldstandard. Die Analyse erfolgt mittels Lichtmikroskopuntersuchung und Immunfluoreszenztest. Bei gleichzeitiger Nieren- und Lungenbeteiligung ist aufgrund der Lebensgefahr bei einer Lungenblutung die Nieren- einer Lungenbiopsie vorzuziehen. Bei Patienten mit erhaltener Nierenfunktion ist diese jedoch weder diagnostisch noch prognostisch von Nutzen. Aufgrund der bei unserem Patienten bronchoskopisch durchgeführten transbronchialen Biopsie wird mittels Immunfluoreszenztest ein Goodpasture-Syndrom mit linearen IgG- und C3-Komplement-Ablagerungen entlang der alveolären Basalmembran bestätigt (Abb. 4).
Abbildung 4: Direkter Immunfluoreszenztest anhand der Lungenbiopsie mit linearen Anfärbungen von IgG-Ablagerungen entlang der alveolären Basalmembran (400-fache Vergrösserung). Vielen Dank an Herrn Dr. med. Samuel Rotman vom Institut für Pathologie für die zur Verfügung gestellte Fotografie.
Die Sensitivität des hochspezifischen (>97%) ELISA-Tests auf anti-GBM-Antikörper im Blut variiert entsprechend des verwendeten Testkits (90,9–100%). In unserem Fall war das Resultat ebenso wie der mittels Elektrochemilumineszenz erhaltene Wert (ECL = 18 CU; normal <20) negativ. Dies ist damit zu erklären, dass die ELISA-Testkits nicht alle für das Goodpasture-Syndrom verantwortlichen Epitope abdecken (siehe Diskussion).
Der direkte Immunfluoreszenztest auf anti-GBM-Antikörper im Blut ist nicht so sensitiv wie die ECL-Methode, weist jedoch eine gute Spezifizität mit unter 10% falsch positiven Resultaten auf. Es ist zu beachten, dass die Menge der zirkulierenden anti-GBM-Antikörper nicht mit der Schwere der Erkrankung korreliert.
Der Western Blot ist eine zusätzliche Methode, um die Resultate des ELISA-Tests zu bestätigen, die in manchen Fällen aufgrund ungereinigter Antigene falsch positiv sind. Er ist nicht überall verfügbar.
Frage 5: Welches ist die auf Grundlage der vorhergehenden Resultate und klinischen Symptome sinnvollste Behandlung?
a) Wiederholte Bluttransfusionen
b) Eine Kortikoidtherapie und Tabakverzicht
c) Eine Kortikoidtherapie und Plasmapheresen
d) Eine Kortikoidtherapie und Cyclophosphamid
e) Eine Kortikoidtherapie, Plasmapheresen und Cyclophosphamid
Eine Unterstützung durch Bluttransfusionen kann, insbesondere bei starken Hämoptysen, erforderlich werden. Diese allein tragen jedoch nicht zur Behandlung der zugrunde liegenden Erkrankung bei.
Die eigentliche Grundsäule der Behandlung des Goodpasture-Syndroms ist eine hochdosierte Kortikoidtherapie, um die Antikörperproduktion zu stoppen und die für ihre Produktion verantwortlichen Zellen zu erreichen. Ein Tabakverzicht ist unerlässlich, um schädliche Reizungen der Atemwege zu vermeiden.
Die Plasmapherese (PEX) ist eine weitere Behandlungssäule, um zirkulierende Antikörper rasch aus dem Blutkreislauf zu eliminieren. Eine Kontrolle des Antikörpertiters ist zur Entscheidung über eine mögliche PEX-Behandlung angezeigt. Nach Absetzen der Plasmapherese muss die Kortikoidtherapie aufgrund des Risikos eines Rebounds der Antikörperproduktion fortgesetzt werden.
Üblicherweise wird der Beginn einer immunsuppressiven Therapie mit Cyclophosphamid empfohlen. Aufgrund dessen starker immunsuppressiver Wirkung sollten vor Behandlungsbeginn, wenn möglich, eine Impfauffrischung und aufgrund des Risikos sekundärer Sterilität bei einem derart jungen Patienten eine Spermaentnahme erfolgen. Eine prophylaktische Behandlung mit Trimethoprim-Sulfamethoxazol zur Prävention einer Lungenentzündung mit Pneumocystis jirovecii wird je nach Fall entschieden.
Bei unserem Patienten wird wegen der diffusen AH, die auf einer zweiten CT-Aufnahme bereits weiter fortgeschritten war, eine Kombinationsbehandlung gewählt. Aufgrund des guten Verlaufs kann die Plasmapherese in unserem Fall nach sechs Sitzungen abgesetzt werden. Die immunsuppressive Behandlung mit Cyclophosphamid wird ambulant drei und die Kortikoidtherapie sechs Monate fortgesetzt (bei unvollständiger Remission nach drei Monaten bis zu neun Monate). Eine therapeutische Alternative mit ebenso schnellem Ansprechen stellt heute in weniger schweren Fällen Rituximab dar.
Diskussion
Das Goodpasture-Syndrom ist eine seltene Autoimmunkrankheit, die erstmals im Jahr 1919 beschrieben wurde. Sie wird auch anti-GBM-Antikörper-Krankheit oder extrakapilläre Glomerulonephritis Typ 1 genannt. Diese rasch progrediente Glomerulonephritis tritt typischerweise in >90% der Fälle auf. In 40–60% der Fälle ist zusätzlich und in 10% der Fälle ausschliesslich die Lunge betroffen. Die erste Manifestation bei einer Lungenbeteiligung sind Hämoptysen, die in manchen Fällen mit trockenem Husten und Dyspnoe einhergehen. Des Weiteren können andere unspezifische Symptome wie Müdigkeit, Schwäche, Übelkeit, Erbrechen, Appetitverlust, Blässe usw. auftreten. In einigen Fällen ist auch das Innenohr mit plötzlichem oder schleichendem Hörverlust betroffen. Von der Erkrankung sind eher 20–30-jährige Männer und 60–70-jährige Frauen betroffen. Obgleich die Ätiopathogenese noch zum Teil unverstanden ist, scheinen genetische Faktoren eine wichtige Rolle zu spielen, da die humanen Leukozytenantigene (HLA) bei der Erkrankung abweichend repräsentiert sind. Die Antikörper sind entweder ausschliesslich gegen eine Monomereinheit der NC1-Domäne der alpha-3-Kette mit einer Molekularmasse von 28 kD oder ausserdem gegen die alpha-4- und -5-Kette des Kollagens Typ IV (alpha3[IV]NC1) gerichtet. Vor Kurzem wurden zudem Antikörper beschrieben, die ausschliesslich gegen die alpha-5-Kette gerichtet sind. Dies trifft auch auf unseren Patienten zu und erklärt das negative Resultat der ELISA-Tests, welche auf die alpha-3-Kette abzielten. Die entsprechenden Epitope sind hauptsächlich in Niere und Lunge zu finden. Für die Exposition der Antikörper gegenüber der Basalmembran der alveolären Kapillaren ist jedoch wahrscheinlich eine vorhergehende alveoläre Läsion erforderlich, was erklären könnte, warum 85% der Betroffenen Raucher sind. Obgleich die Erkrankung früher sehr häufig tödlich verlief, beträgt ihre heutige 5-Jahres-Überlebensrate dank der Kombinationsbehandlung aus Plasmapherese, Kortikoiden und Immunsuppressiva 80%.
Antworten:
Frage 1: e. Frage 2: c. Frage 3: b. Frage 4: a. Frage 5: e.
Disclosure statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Korrespondenz
Dr. med. Saif Al Jarrah Service de médecine interne Centre Hospitalier Universitaire Vaudois Rue du Bugnon 46 | BH10-670 CH-1011 Lausanne saif_aljarrah[at]me.com
Literatur
1 Manzoni R. Fitting JW, Bart PA. Approche diagnostique devant une hémoptysie: à propos d’un cas. Swiss Med Forum. 2014;14(11).
2 Hié M. Syndrome pneumo-rénal: une urgence diagnostique et thérapeutique pour le réanimateur et l’interniste. Revue de médecine interne. 2013:679–86.
4 Lazor R. Alveolar hemorrhage in anti-basement membrane antibody disease: a series of 28 cases. Medicine (Baltimore). 2007:181–93.
5 Greco A. Goodpasture’s syndrome: a clinical review. Autoimmunity reviews. 2015:246–53.
6 Cui T, et al. Antibodies to α5 chain of collagen IV are pathogenic in Goodpasture’s disease. J Autoimmun. 2016:1–11.
7 Sinico RA, Radice A, Corace C, et al. Anti-glomerular basement membrane antibodies in the diagnosis of Good Pasture syndrome: a comparison of different assays. Nephrol Dial Transplant. 2006:397–401.
8 Pedchenko V, Bondar O, Fogo AB, et al. Molecular architecture of the Goodpasture autoantigen in anti-GBM nephritis. N Engl J Med. 2010:343–54.
9 Giulieri S, Tissot F. Les spirochètes dans tous leur états. Rev Med Suisse. 2010:721–6.
10 Pardon A, Aubert A, Bart PA. Biomarqeurs en immunologie générale. Rev Med Suisse. 2013;1982–91.