a Anatomisches Institut der Universität Zürich; b Abteilung für Neurologie, Stadtspital Triemli, Zürich; c Universitätsinstitut für Diagnostische und Interventionelle Neuroradiologie, Inselspital Bern; d Universitätsklinik für Rheumatologie, Immunologie und Allergologie, Inselspital Bern; e Institut für Pathologie, Universität Bern
Die Zuweisung der 58-jährigen Patientin erfolgte durch eine niedergelassene Neurologin zur weiteren ätiologischen Zuordnung einer Polyneuropathie.
Hintergrund
Eine Polyneuropathie eröffnet zumeist eine breite ätiologische Differentialdiagnose. Je älter die Patienten, desto häufiger gelingt die Ursachenklärung trotz hohem diagnostischem Aufwand nicht. Der folgende Fall demonstriert, dass es sich lohnt, hellhörig zu sein und auf feine Hinweise wie beispielsweise Sicca-Beschwerden zu achten.
Fallbericht
Anamnese
Die Zuweisung der 58-jährigen Puppenmacherin erfolgte durch eine niedergelassene Neurologin zur weiteren ätiologischen Zuordnung einer Polyneuropathie. Die Patientin bemerkte ein Jahr vor Präsentation eine Gefühlsstörung in den Zehen beidseits mit langsamer Ausbreitung bis zu den Knien. Drei Wochen vor Hospitalisation kam es zu einer schmerzlosen, akuten Verschlechterung im Sinne eines Kraftverlustes in den Beinen sowie einer Feinmotorikstörung und Hypästhesie beider Hände. Arbeiten als Puppenmacherin war nicht mehr und Treppensteigen nur noch auf allen Vieren möglich. Auf Nachfrage berichtete die während der Anamnese immer wieder zur Wasserflasche greifende Patientin, seit Jahren unter einer Xerostomie und Xerophthalmie sowie einem Raynaud-Syndrom zu leiden. B-Symptome oder Gelenkbeschwerden wurden verneint.
Status
Es fand sich eine rechts und distal betonte schlaffe Tetraparese (Kraftgrad gemäss «Medical Research Council» für Fussdorsalextension beidseits M1) sowie eine distal betonte Hypästhesie der Unterarme und Unterschenkel für alle Qualitäten mit Akzentuierung im Versorgungsgebiet der Nervi ulnares bei schwachen bis fehlenden Muskeleigenreflexen.
Befunde
Elektrophysiologisch liess sich eine schwere, axonale sensomotorische Polyneuropathie nachweisen. Im Saxon- und Schirmer-Test konnte die geschilderte Sicca-Symptomatik objektiviert werden.
Die laborchemischen Auffälligkeiten sind in Tabelle 1 aufgeführt. Neben einem inflammatorischen Syndrom fanden sich positive ANA, SSA- und SSB-Antikörper sowie eine polyklonale Hypergammaglobulinämie mit stark erhöhten Leichtketten. Bei positivem Rheumafaktor mit Komplementverbrauch konnten Kryoglobuline vom Typ II (monoklonales IgM kappa sowie polyklonales IgG) mit einem Kryokrit von 5,4% detektiert werden. Ein Suchtest auf Hepatitis C blieb negativ. Der Liquor wies liquorspezifische oligoklonale Banden bei sonst unauffälligen Werten auf, zusätzlich waren identische Banden in Liquor und Serum vorhanden. In der Lippenspeicheldrüsenbiopsie imponierte eine ausgeprägte chronische lymphozytäre Entzündung mit interstitieller Fibrose ohne vermehrt IgG4-positive Plasmazellen. Der Focus-Score belief sich auf >1 (d.h. Nachweis von mehr als einem lymphozytären Fokus bestehend aus mindestens 50 Lymphozyten pro 4 mm2). In der Knochenmarksbiopsie betrug die Plasmazellvermehrung <10%. Bei Nachweis einer klonalen Plasmazellpopulation mit Leichtkettenrestriktion vom Typ IgM kappa in der Durchflusszytometrie waren die Befunde vereinbar mit einer monoklonalen Gammopathie unklarer Signifikanz (MGUS).
Tabelle 1: Laborresultate der Patientin bei Eintritt.
Patientin
Norm
Erythrozytensedimentationsrate (ESR) (mm/h)
94
<30
Kappa (g/l)
472,5
3,3–19,4
Lambda (g/l)
156,5
5,7–26,3
Kappa/Lambda
3,02
0,26–1,65
Gamma-Globulin (g/l)
35,8
7,7–14,6
Rheumafaktor (IU/ml)
355
<20
ANA (Titer)
>1:1280
<1:80
SSA/Ro (Units)
106
<20
SSB/La (Units)
132
<20
C3 (g/l)
0,77
0,9–1,8
C4 (g/l)
0,08
0,1–0,4
Verlauf und Therapie
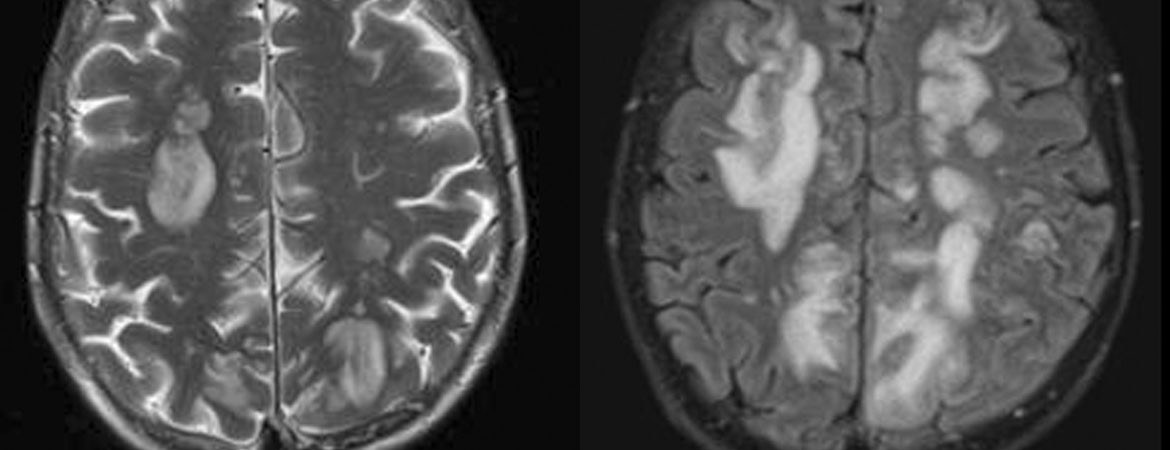
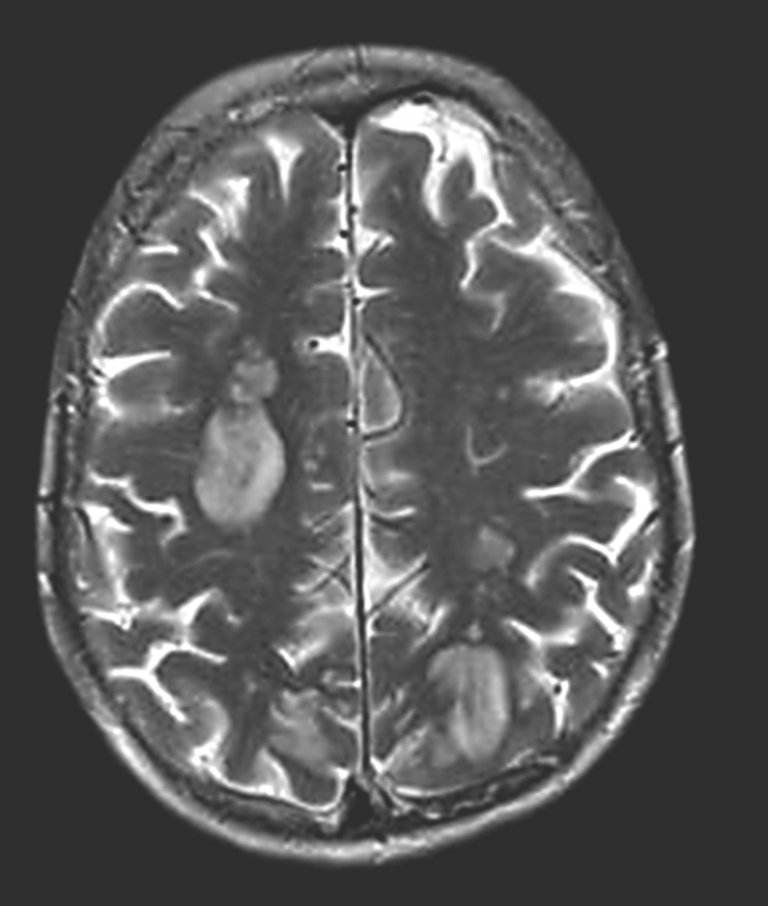
Einen Tag nach Beginn einer intravenösen Glukokortikoid-Stosstherapie (Methylprednisolon [Solu-Medrol®] 500 mg) erlitt die Patientin einen erstmaligen generalisierten epileptischen Anfall. In der Magnetresonanztomographie (MRT) des Schädels fanden sich supratentoriell multiple kortikale und subkortikale T2-gewichtet(T2w)- und Tirm-hyperintense Läsionen frontal, parieto-okzipital und okzipito-temporal beidseits, die nach Gadoliniumgabe eine Kontrastmittelaufnahme zeigten (Abb. 1 zeigt exemplarisch die T2w- Darstellung). Die Glandula parotis stellte sich beidseits vergrössert dar.
Abbildung 1: Initiale Magnetresonanztomographie des Schädels mit disseminiert bihemisphärischen supratentoriellen multiplen kortikalen und subkortikalen T2w-hyperintensen Läsionen.
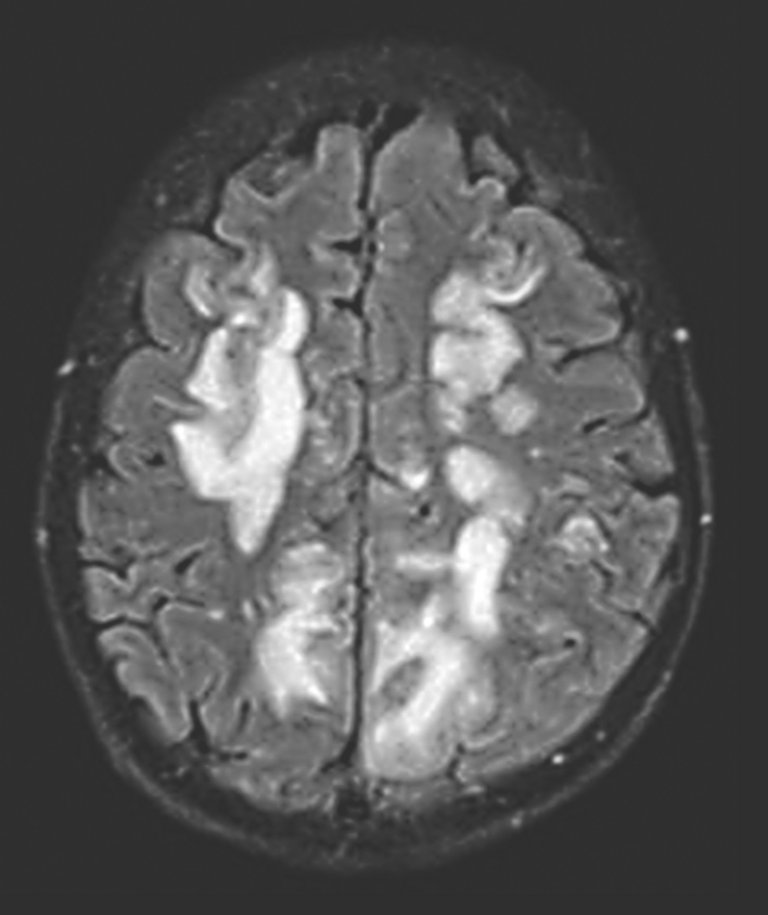
Eine anfallsunterdrückende Therapie mit Levetiracetam wurde initiiert und die intravenöse Steroiddosis verdoppelt. Aufgrund der Beteiligung des Zentralnervensystems (ZNS) erfolgte zusätzlich die Gabe von Rituximab. 24 Stunden nach Verabreichung traten zwei weitere generalisierte Anfälle auf, nach welchen sich die Patientin somnolent zeigte und eine Hemianopsie, einen multimodalen Neglekt nach rechts sowie eine proximale Armparese rechts aufwies. MR-bildgebend fand sich korrelierend zur Klinik eine fulminante Befundverschlechterung mit deutlicher Grössenzunahme der nun flächig konfluierenden Tirm- und T2w-hyperintensen Signalalterationen, insbesondere in den Gyri frontales superior et medius beidseits sowie okzipital links (Abb. 2 zeigt exemplarisch die Tirm-Aufnahme). Neu wiesen insbesondere die Läsionen okzipital und temporal kleine Hämorrhagien auf. Der histologische Befund der Hirnbiopsie konnte weder einer Vaskulitis noch einer Lymphominfiltration zugeschrieben werden.
Abbildung 2: MR-Verlaufsbildgebung mit fulminanter Befundverschlechterung bei deutlicher Grössenzunahme der nun flächig konfluierenden Tirm-hyperintensen Signalalterationen bihemisphärisch supratentoriell.
Im Verlauf von insgesamt fünf Plasmapherese-Sitzungen besserte sich der Allgemeinzustand der Patientin rasch. Ein Cyclophosphamidstoss (750 mg/m2) wurde appliziert, die Glukokortikoide per os verabreicht und sukzessive reduziert. Nach der letzten Plasmapherese liess sich MR-tomographisch kein Kontrastmittel-Enhancement mehr nachweisen. Bei persistierender ausgeprägter schlaffer Tetraparese mit nur am Eulenburg-Gehwagen mobiler und bei der Körperpflege auf Hilfe angewiesener Patientin erfolgte die Entlassung in die stationäre Neurorehabilitation.
Nach bisher siebenmaliger Cyclophosphamidgabe in monatlichen Abständen ist die Patientin ohne Hilfsmittel eine Stunde in normalem Schritttempo gehfähig. Die Feinmotorik der Hände hat sich leicht verbessert, sodass sie mittlerweile dank eigens konstruierter Hilfsmittel wieder in der Lage ist, Handpuppen anzufertigen.
MR-tomographisch ist eine vollständige Regredienz der flächigen kortikalen und subkortikalen bihemisphärischen T2w- und Tirm-hyperintensen Signalalterationen zu verzeichnen. Die Neurographien zeigten eine im Vergleich zur Voruntersuchung stabile schwere sensomotorische axonale Polyneuropathie. Kryoglobuline liessen sich nicht mehr nachweisen. Die MGUS wird durch die Kollegen der Hämatologie engmaschig kontrolliert.
Geplant ist eine Erhaltungstherapie mit Rituximab in 6-monatlichen Abständen.
Diagnose
In Zusammenschau der Befunde stellten wir die Diagnose eines primären Sjögren-Syndroms mit schwerem extraglandulärem Befall im Sinne einer Beteiligung des peripheren und zentralen Nervensystems am ehesten auf dem Boden einer kryoglobulinämischen Vaskulitis. Es bleibt offen, inwieweit die Gabe von Rituximab zur Präzipitation von Immunkomplexen mit den Kryoglobulinen und somit zur rapiden Verschlechterung beitrug. Die klinische Stabilisierung unter Plasmapherese führen wir auf das Auswaschen der somit wahrscheinlich pathogenetisch bedeutsamen Kryoglobuline und/oder Immunkomplexe zurück. Die MGUS werten wir als Folge der chronischen B-Zell- Aktivierung im Rahmen der Grunderkrankung.
Diskussion
Das Sjögren-Syndrom (SS) ist eine autoimmune Erkrankung, die sich klassischerweise mit einer Sicca-Symptomatik bei Frauen um das fünfzigste Lebensjahr manifestiert. Die Prävalenz in der Gesamtbevölkerung wird auf 0,5% geschätzt, womit das SS nach der rheumatoiden Arthritis (RA) die zweithäufigste rheumatologische Krankheit darstellt [1].
Erstmals beschrieb der schwedische Ophthalmologe Henrik Sjögren 1933 die klinischen und histologischen Charakteristika der «Keratokonjunktivitis sicca» in Abgrenzung zur mit einem Vitamin-A-Mangel assoziierten Xerophthalmie.
Das primäre (pSS) wird von einem sekundären SS im Rahmen einer zusätzlichen rheumatologischen Grunderkrankung – zumeist einer RA, einer Systemsklerose oder eines systemischen Lupus erythematodes – abgegrenzt.
Diagnostik
Obwohl primär für Studienzwecke entworfen, können die zuletzt 2016 angepassten internationalen Klassifikationskriterien des «American College of Rheumatology» (ACR) und der «European League against Rheumatism» (EULAR) zur Diagnosestellung herangezogen werden. Sie umfassen unter anderem Resultate einer Speicheldrüsenbiopsie, das Vorliegen von SSA-Antikörpern sowie Tests zur Objektivierung eines verminderten Tränen- und Speichelflusses. Das Vorliegen von Sicca-Symptomen ist bei immunserologischem oder histologischem Nachweis einer Autoimmunreaktion nicht obligat. Medikamente mit Einfluss auf die Salivation und Lakrimation sind 24 Stunden vor Durchführung der objektiven Testung zu pausieren. Infektionen mit dem Hepatitis-C- und HI-Virus, Lymphome, die Sarkoidose sowie IgG4-assoziierte Erkrankungen stellen wichtige Differentialdiagnosen dar und sollten serologisch sowie in der Speicheldrüsenbiopsie gesucht werden [1]. Anlässlich letzterer kann der Focus-Score (siehe oben) zur Quantifizierung der Sialadenitis bestimmt werden. Ein Wert ≥1 gilt als pathologisch, ist jedoch nicht pathognomonisch für ein pSS.
Klinik
Bis zu ⅓ der Patienten leiden unter extraglandulären Manifestationen, die sich zumeist in Form von Arthralgien, einem kutanen Befall, einer pulmonalen Beteiligung oder einer Affektion des peripheren Nervensystems äussern. Das Spektrum der Neuropathien ist breit und reicht von der schmerzhaften «small fibre»-Neuropathie über eine Affektion der Spinalganglien mit sensibler Ataxie bei eingeschränkter Propriozeption bis hin zur Mononeuritis multiplex. Axonale sensomotorische Polyneuropathien – wie bei unserer Patientin – sind mit einer Kryoglobulinämie und einem erhöhten Lymphomrisiko assoziiert [2, 3]. Unklar bleibt, inwieweit es sich um T-Zell-mediierte Vaskulitiden der Vasa nervorum oder um Immunkomplex-vermittelte Immunreaktionen handelt, wobei je nach Phänotyp unterschiedliche Mechanismen involviert zu sein scheinen [2].
In der Literatur finden sich divergierende Angaben zur Häufigkeit und Art einer ZNS-Beteiligung bei pSS, wofür neben einem «referral bias» die variable Klinik sowie wechselnde und heterogene Klassifikationskriterien verantwortlich sein dürften. Beschrieben sind fokale und disseminierte T2w-hyperintese zerebrale und spinale Läsionen, die an Demyelinisierungsherde ähnlich einer Multiplen Sklerose erinnern. Eine Assoziation des pSS mit der Neuromyelitis-optica-Spektrum-Störung ist beschrieben. Man geht von einer hierfür verantwortlichen Prädisposition für humoral vermittelte Autoimmunreaktionen aus [3].
Komplikationen
Gefürchtete Komplikationen des SS als Folge einer chronischen polyklonalen B-Zell-Aktivierung sind die Entwicklung lymphoproliferativer Neoplasien sowie das Auftreten einer kryoglobulinämischen Vaskulitis.
Das relative Risiko für B-Zell-Non-Hodgkin-Lymphome – allen voran MALT-Lymphome (Lymphome des mukosa-assoziierten lymphatischen Gewebes) der Speicheldrüsen – gegenüber der Allgemeinbevölkerung ist bis zu 40-fach erhöht. Regelmässige klinische, laborchemische und gegebenenfalls sonographische Kontrollen sind indiziert, wobei keine Guidelines bezüglich Art und Häufigkeit des Screenings existieren [1].
Kryoglobuline sind Immunglobuline, die in vitro bei niedrigen Temperaturen präzipitieren [4]. In Kohortenstudien lassen sich bei 5–15% der an einem pSS Erkrankten Kryoglobuline nachweisen, wobei hiervon rund 50% eine kryoglobulinämische Vaskulitis entwickeln [2, 4]. Meist finden sich beim pSS Kryoglobuline vom Typ II, das heisst monoklonale IgM- mit polyklonalen IgG-Antikörpern. Die IgM-Antikörper verfügen über die Fähigkeit, den konstanten Teil des IgGs zu binden, womit sie Rheumafaktoraktivität aufweisen. Der nach dem Erstbeschreiber als Meltzer-Trias bekannte Symptomkomplex aus palpabler Purpura, Arthralgien und allgemeiner Schwäche wird in 80% der Fälle einer kryoglobulinämischen Vaskulitis beobachtet [4].
Therapie
Die Therapie des pSS richtet sich nach dem Befallsmuster. Bei rein glandulärer Beteiligung reicht oftmals eine symptomatisch-topische Therapie (Tränenersatzmittel, Anregung des Speichelflusses mittels Bonbons/Kaugummis). Auf eine gute Zahnhygiene ist zu achten. Die Behandlung bei extraglandulärem Befall basiert vornehmlich auf Expertenmeinungen. Bei Neuropathien werden in der Akutphase Steroide oder intravenöse Immunglobuline eingesetzt; Azathioprin und Mycophenolat Mofetil kommen als Erhaltungstherapie zum Einsatz. Zunehmend findet Rituximab aufgrund der pathogenetischen Bedeutung der B-Zellen Verwendung. Die Art der Polyneuropathie scheint das Therapieansprechen auf die Immunsuppressiva und -modulatoren zu beeinflussen [2, 3]. Im Falle einer kryoglobulinämischen Vaskulitis mit fulminantem Verlauf empfiehlt sich eine Plasmapherese. Im Anschluss kommen Cyclophosphamid und Rituximab zum Einsatz [4]. In Zusammenhang mit der Verabreichung von Rituximab bei Hepatitis-C-Virus-assoziierter gemischter kryoglobulinämischer Vaskulitis wurden bei sechs von 22 Patienten schwere systemische Reaktionen 1–9 Tage nach Verabreichung von Rituximab beschrieben. Als mögliche Erklärung wird die Immunkomplexbildung zwischen dem gegen CD20 gerichteten monoklonalen Antikörper und den Kryoglobulinen genannt [5]. Dies könnte die rapide klinische Verschlechterung unserer Patientin nach Rituximaberstgabe erklären. Weitere Daten zu dieser Beobachtung fehlen.
Das Wichtigste für die Praxis
• Sicca-Symptome sind häufig und nicht immer auf ein Sjögren-Syndrom (SS) zurückzuführen; im Zweifelsfall empfiehlt sich eine Schirmer- und Saxon-Testung sowie eine Biopsie der Lippenspeicheldrüsen.
• Neben einem Befall der Speichel- und Tränendrüsen ist ein extraglandulärer Befall mit Arthralgien, Beteiligung der Haut, Lungen sowie des peripheren und selten des zentralen Nervensystems möglich.
• Bei bis zu 15% der Patienten mit primärem SS (pSS) lassen sich Kryoglobuline nachweisen, die in rund der Hälfte zu einer kryoglobulinämischen Vaskulitis führen. Besonders bei Nachweis eines Rheumafaktors sowie einem Komplementverbrauch sollte hieran gedacht werden. Diese sind mit einem schweren systemischen Befall und erhöhtem Lymphomrisiko assoziiert.
• Patientinnen mit pSS weisen ein erhöhtes Risiko für die Entwicklung lymphoproliferativer Neoplasien (v.a. MALT-Lymphome als auch Plasmazellneoplasien) auf; regelmässige Screening-Untersuchungen sind daher indiziert (Suche nach Lymphadenopathie, Sonographie der Parotis, Eiweisselektrophorese mit Immunfixation, Laktatdehydrogenase und β2-Mikroglobulin).
• Die Therapie richtet sich nach klinischem Befallsmuster und Schweregrad und reicht von rein symptomatisch-topischen Therapeutika bis hin zur Plasmapherese. Insbesondere bei schwerer systemischer Krankheitsmanifestation ist die Datenlage bezüglich optimaler Behandlung beschränkt.
Disclosure statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Korrespondenz
Dr. med. Kathi Ging Anatomisches Institut der Universität Zürich Winterthurerstrasse 190 CH-8057 Zürich kathi.ging[at]anatomy.uzh.ch
Literatur
1 Fox RI. Sjögren’s Syndrome. Lancet. 2005;366(9482):321–31.
2 Pavlakis PP, Alexopoulos H, Kosmidis ML, Mamali I, Moutsopoulos HM, Tzioufas AG, et al. Peripheral neuropathies in Sjögren’s Syndrome: A critical update on clinical features and pathogenic mechanisms. J Autoimmun. 2012;39(1-2):27–33.
3 Berkowitz AL, Samuels MA. The neurology of Sjögren’s syndrome and the rheumatology of peripheral neuropathy and myelitits. Pract Neurol. 2014;14(1):14–22.
4 Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinemias. Lancet. 2012;379(9813):348–60.
5 Sène D, Ghillani-Dalbin P, Amoura Z, Musset L, Cacoub P. Rituximab may form a complex with IgMkappa mixed Cryoglobulin and induce severe systemic reactions in patients with Hepatitis C Virus-induced Vasculitis. Arthritis Rheum. 2009;60(12):3848–55.