1 Frederiksen H, Schmidt K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood. 1999;94(3):909.
2 Terrell DR, Beebe LA, Vesely SK, Neas BR, Segal JB, George JN. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol. 2010;85(3):174.
3 Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol. 2009;83(2):83–9. Epub 2009 Feb 23.
4 Terrell DR, Beebe LA, Neas BR, Vesely SK, Segal JB, George JN. Prevalence of primary immune thrombocytopenia in Oklahoma. Am J Hematol. 2012. Sep;87(9):848–52. Epub 2012 Jun 5.
5 Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood. 2001;97(9):2549.
6 Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR, Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol. 2003;122(6):966.
7 Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386.
8 Lambert M, Gernscheimer T. Clinical updates in adult immune thrombocytopenia. Blood. 2017;129:2829–35.
9 Swinkels M, Rijkers M, Voorberg J, Vidarsson G, Leebeek FWG, Jansen AJG. Emerging Concepts in Immune Thrombocytopenia. Front. Immunol. 2018;9:880.
10 Stasi R, Provan D. Helicobacter pylori and Chronic ITP. Hematology Am Soc Hematol Educ Program. 2008.
11 Audia S, Mahévas M, Samson M, Codeau B, Bonnotte B. Pathogenesis of immune thrombocytopenia. Autoimmunity Reviews. 2017;16:620–32.
12 Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood. 2009;113(26):6511. Epub 2009 Apr 24.
13 Mahévas M, Chiche L, Uzunhan Y, Khellaf M, Morin AS, Le Guenno G, Péronne V, et al. Association of sarcoidosis and immune thrombocytopenia: presentation and outcome in a series of 20 patients. Medicine (Baltimore). 2011;90(4):269.
14 Cines DB, Liebman H, Stasi R. Pathobiology of secondary immune thrombocytopenia. Semin Hematol. 2009;46(1 Suppl 2):S2.
15 Neunert C, Noroozi N, Norman G, Buchanan GR, Goy J, Nazi I, et al. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J Thromb Haemost. 2015;13(3):457. Epub 2015 Jan 14.
16 Neunert C, Lim W, Crowther M, Cohen A, Solberg L Jr, Crowther MA. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117(16):4190.
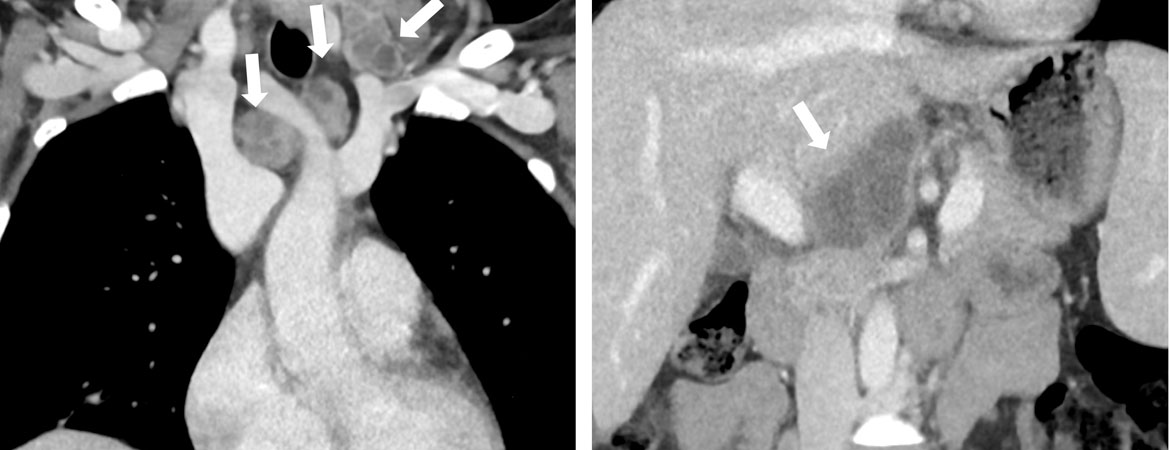
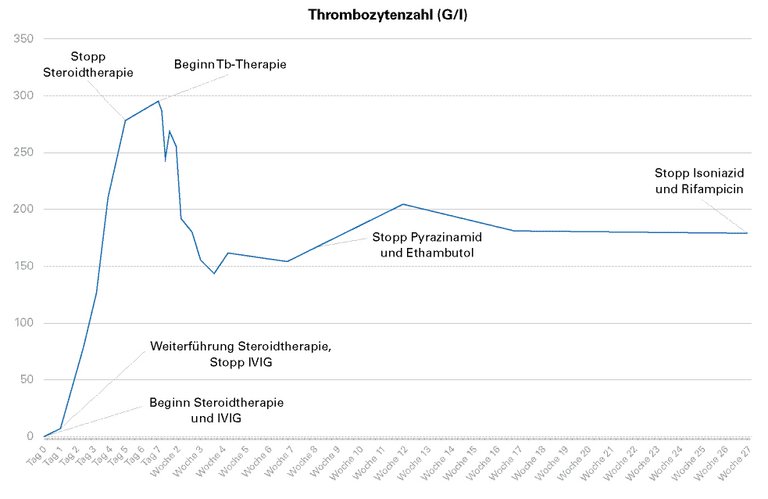
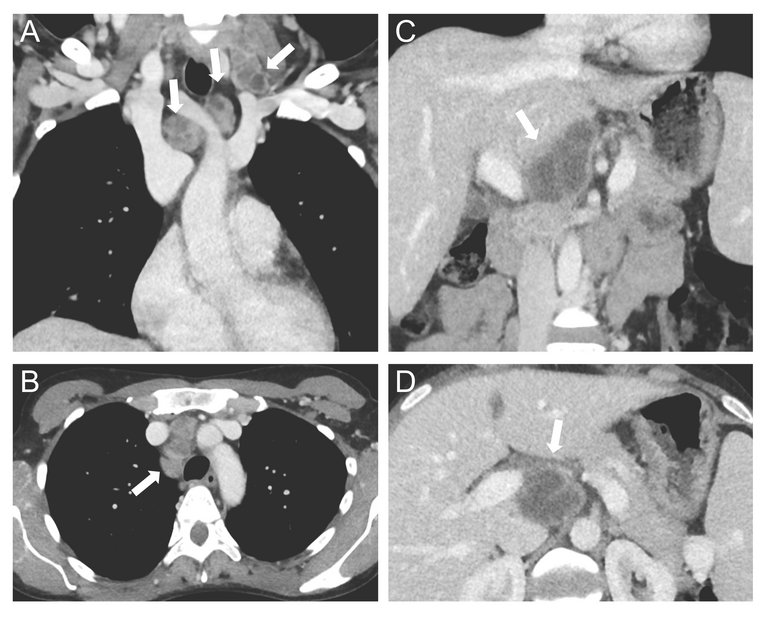
17 Weber S, Bélard S, Rai S, Reddy S, Belurkar S, Saravu S. Immune thrombocytopenia secondary to tuberculosis: a case and review of literature. Int J Tuberc Lung D. 2017;21(4):466–70.
18 Shu-Ruei Wu, Hsiao-Ching Kuo. Awareness of tuberculosis among patients with newly diagnosed immune thrombocytopenia in the endemic area. Ann Hematol. 2017;96:1773–4.
19 Ramagopalan S, Goldacre R, Skingsley A, Conlon C, Goldcare M. Associations between selected immune-mediated diseases und tuberculosis: record-linkage stuidies. BMC Medicine. 2013;11:97.
20 Cooper N. State of the art – how I manage immune thrombocytopenia. British Journal of Hematology. 2017;177:39–54.
21 Neunert C. Management of newly diagnosed immune thrombocytopenia: can we change outcomes? Blood advances. 2017;1(24):2295–301.
22 Zaja F, Bacarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood. 2010;115(14):2755–62.