Eine 65-jährige Patientin mit langjährig bekannter idiopathischer vasoreaktiver pulmonal-arterieller Hypertonie präsentierte sich mit akuter Dyspnoe und Thoraxschmerzen auf einer Notfallstation.
Hintergrund
Wir berichten über eine 65-jährige Patientin mit langjährig bekannter idiopathischer vasoreaktiver pulmonal-arterieller Hypertonie und akuter Dissektion der Pulmonalarterie. Dies führte zur Kompression der Koronararterien mit konsekutiv ausgedehntem Myokardinfarkt und kardiogenem Schock. Trotz notfallmässigem Graft-Ersatz der Pulmonalarterie kam es in der Folge zum Multiorganversagen, und die Patientin verstarb. Eine Dissektion der Pulmonalarterie ist eine seltene, potentiell hochletale Komplikation bei Patienten mit idiopathischer pulmonal-arterieller Hypertonie.
Fallbericht
Anamnese
Vor 30 Jahren wurde bei der Patientin erstmalig eine idiopathische pulmonal-arterielle Hypertonie (IPAH) diagnostiziert. In der Pulmonalisangiographie zeigte sich bereits damals eine erhebliche Dilatation des Truncus pulmonalis. Eine chronisch thromboembolische pulmonale Hypertonie konnte angiografisch ausgeschlossen werden. Bei damals fehlenden Therapiemöglichkeiten wurde keine spezifische Behandlung eingeleitet.
2005 wurde die Patientin erstmalig in der Spezialsprechstunde für Pulmonale Hypertonie beurteilt. In der Rechtsherzkatheteruntersuchung bestätigte sich eine schwere prä-kapilläre pulmonale Hypertonie (mittlerer pulmonal-arterieller Druck (mPAP) 55 mm Hg [Norm <25 mm Hg], Wedge-Druck 6 mm Hg [Norm ≤15 mm Hg], Cardiac-Index 2,9 l/min/m2 [Norm 2,4–4 l/min/m2], pulmonal vaskulärer Widerstand [PVR] 12 Wood-unit [Norm <3 WU]). Bei erfüllten Vasoreaktivitäts-Kriterien wurde die Diagnose einer vasoreaktiven IPAH gestellt und eine Therapie mit einem Kalzium-Antagonisten («calcium channel blocker» [CCB]) eingeleitet. Wegen Beinödemen, welche die Patientin sehr störten, konnte die CCB-Therapie nicht adäquat gesteigert werden. Zusätzlich wurde die persistierende belastungsabhängige Dyspnoe daher ab 2012 mit einem Endothelin-Rezeptor-Antagonisten (Bosentan) behandelt. Darunter kam es zu einer Stabilisierung der Symptome (NYHA II), die Patientin war im Alltag weitgehend beschwerdefrei, hatte eine normale Sechs-Minuten-Gehstrecke von 632 Metern mit leichter Entsättigung unter Belastung auf 88% und ein nur leicht erhöhtes NT-pro-BNP von 395 ng/l.
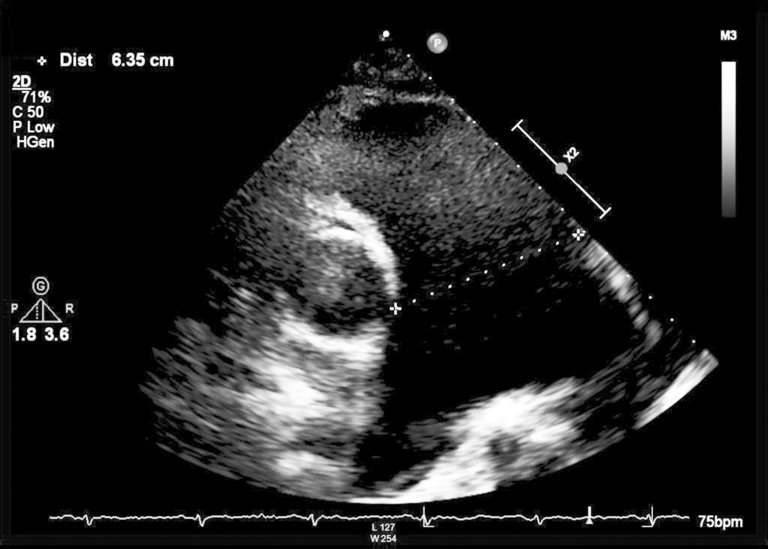
Die zuletzt durchgeführte Echokardiographie aus dem Jahr 2017 (Abb. 1) zeigte eine deutliche persistierende Dilatation des Truncus pulmonalis mit einem Durchmesser von 6,4 cm, die seit mindestens 2013 als stabil dokumentiert war, sowie eine rechtsventrikuläre Dilatation mit erhaltener systolischer rechtsventrikulärer Globalfunktion.
Abbildung 1: Transthorakale Echokardiographie von 2017, parasternale kurze Achse über die Aortenklappe (Bildmitte). Der Truncus pulmonalis (rechts) ist deutlich dilatiert (>6 cm).
Status und Befunde
Im Anschluss an einen grippalen Infekt der oberen Luftwege präsentierte sich die Patientin Anfang 2018 mit akuter Dyspnoe und Thoraxschmerzen auf einer Notfallstation. Bei klinischer und radiologischer Diagnose eines Lungenödems und rasch zunehmender respiratorischer und hämodynamischer Verschlechterung musste die Patientin intubiert werden. Im Elektrokardiogramm imponierten ST-Hebungen in aVR und diffuse ST-Senkungen als Hinweis für eine globale Ischämie (Abb. 2).
Abbildung 2: Elektrokardiogramm beim Eintritt auf die Notfallstation. Sinusrhythmus mit diffusen ST-Senkungen, ST-Hebung in aVR.
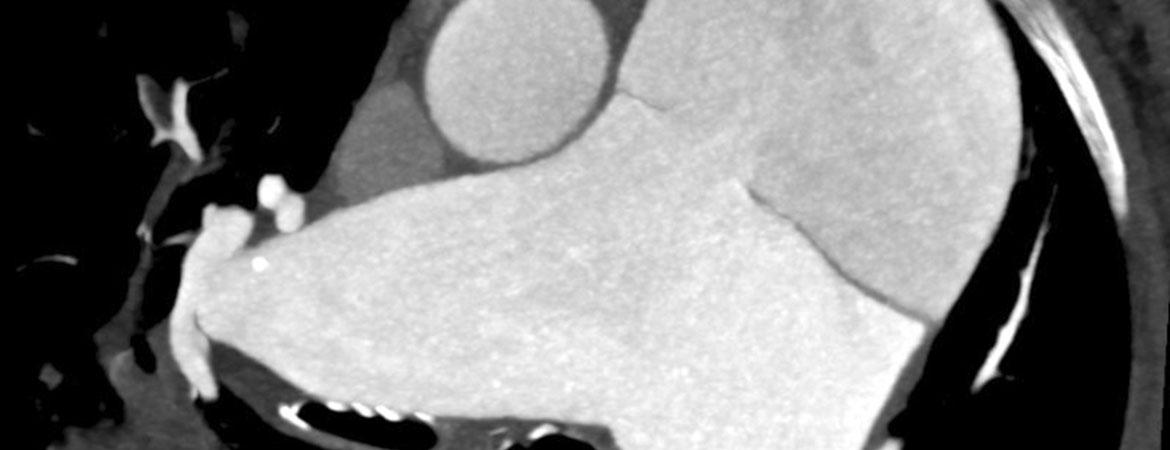
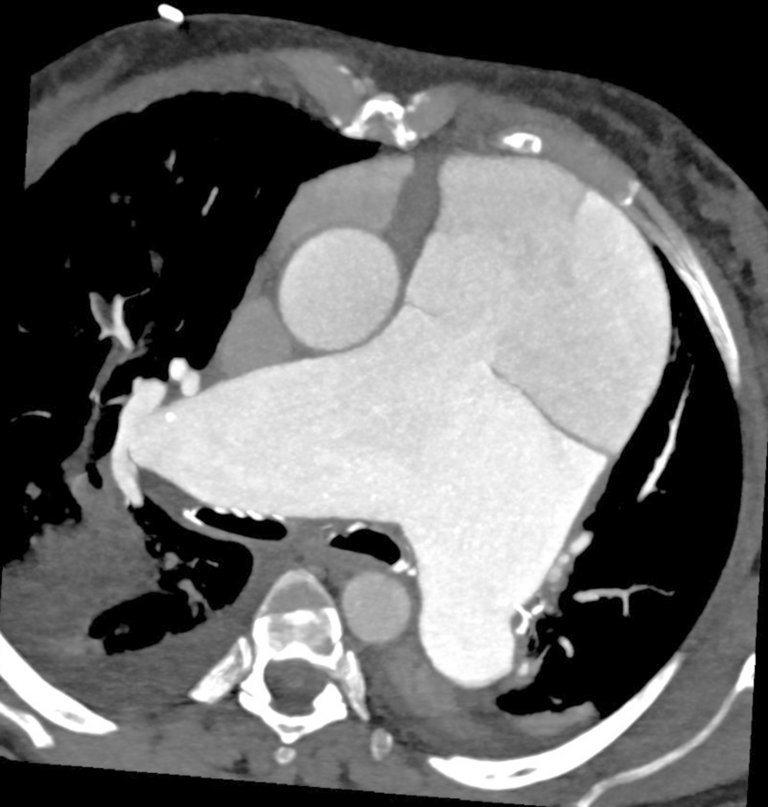
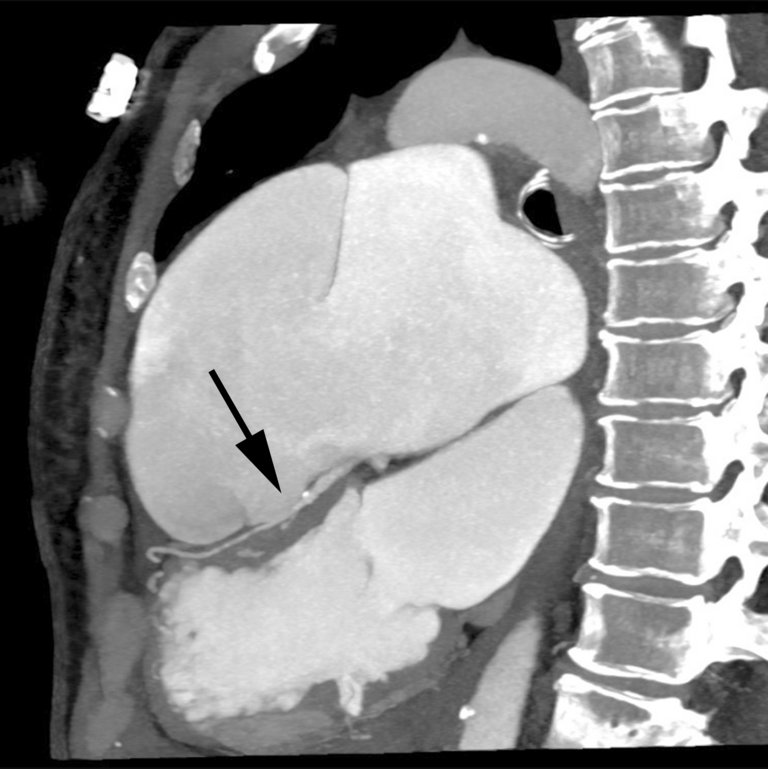
In der Computertomographie kam eine Dissektion des Truncus pulmonalis bis in die rechte Pulmonalarterie zur Darstellung mit Kompression des grosskalibrigen proximalen Ramus interventricularis anterior (RIVA) und des proximalen Ramus circumflexus, die beide mit separaten Ostien aus der Aorta abgingen (Abb. 3, 4 und 5).
Abbildung 3: Kontrastmittelverstärkte Computertomographie-Pulmonalisangiographie: axiale dünne «maximum intensity projection» (MIP) mit Darstellung der massiven Dilatation des Truncus pulmonalis und der Pulmonalarterien sowie der Dissektionsmembran.
Abbildung 4: Kontrastmittelverstärkte Computertomographie-Pulmonalisangiographie: 3D-Darstellung des Aneurysmas sowie der Dissektion mittels «cinematic rendering».
Abbildung 5: Kontrastmittelverstärkte Computertomographie-Pulmonalisangiographie: koronare dünne «maximum intensity projection» (MIP) mit Darstellung der Kompression des kaliberkräftigen mittleren RIVA (Pfeil) durch das Aneurysma und Dissektion des Truncus pulmonalis.
Therapie und Verlauf
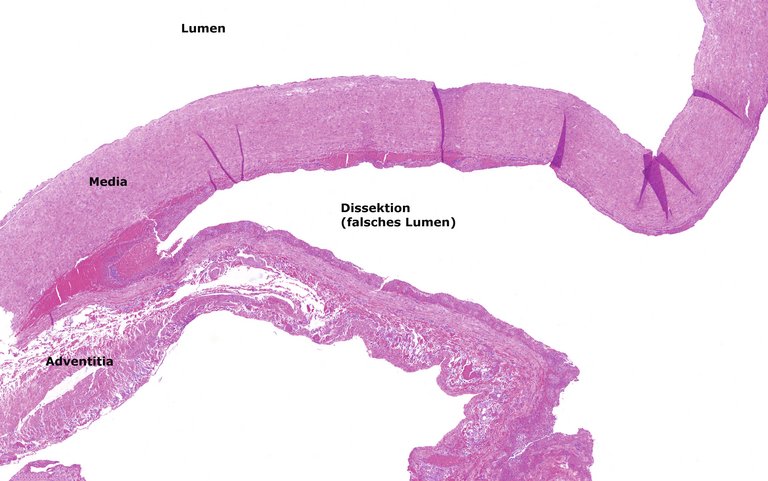
Die Patientin wurde umgehend notfallmässig operiert. Es erfolgte eine klappenerhaltende Resektion des Truncus pulmonalis und der rechten Pulmonalarterie mit Ersatz durch einen Dacron-T-Graft 28 mm. Histologisch bestätigte sich die Dissektion im äusseren Mediadrittel mit Bildung eines falschen Lumens (Abb. 6).
Abbildung 6: Pulmonalarteriendissektion: Spaltung und Einblutung in die muskuläre Gefässwand innerhalb des äusseren Mediadrittels. Geringe Fibrose der Adventitia. (Färbung: Hämatoxylin und Eosin, Vergrösserung: 500 μm).
Postoperativ präsentierte sich die Patientin in kombiniert kardiogenem und distributivem Schock mit erhöhtem Bedarf an Inotropika und Vasopressoren. Aufgrund einer weiteren hämodynamischen Verschlechterung musste eine periphere venoarterielle extrakorporale Lebenserhaltung (vaECLS) etabliert werden. In der Folge entwickelten sich eine schwere Hämoptoe sowie eine Niereninsuffizienz und ein Multiorganversagen, woran die Patientin zwölf Tage später verstarb.
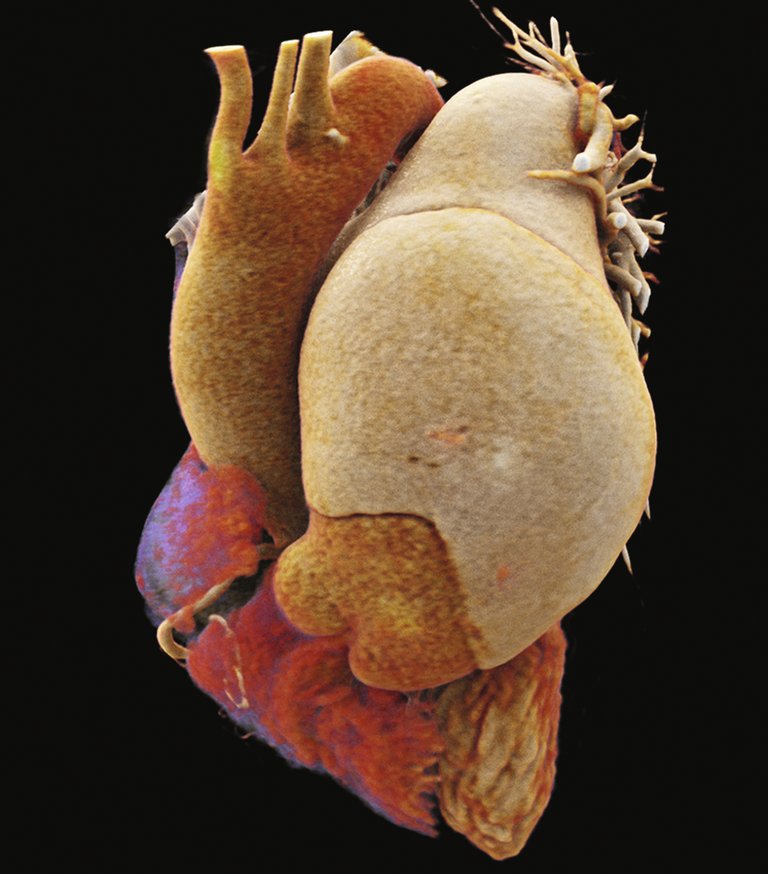
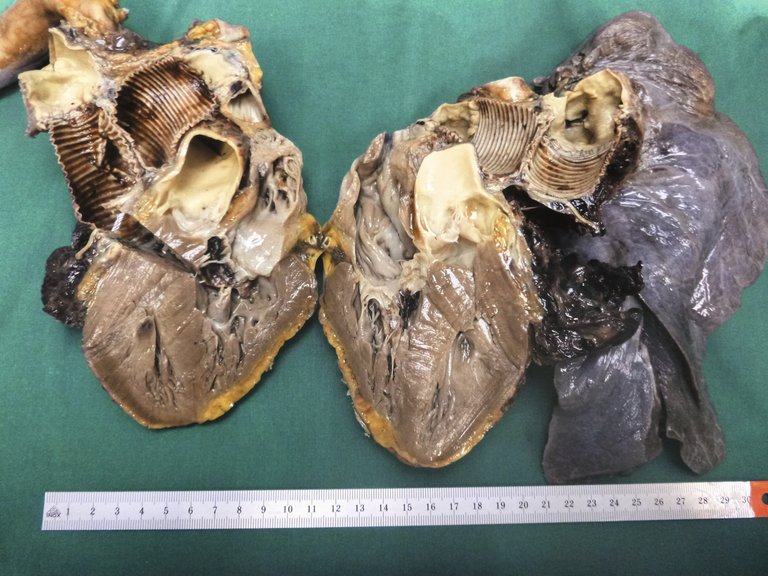
In der Autopsie kam ein ausgedehnter subakuter Myokardinfarkt mit Beteiligung der linken Vorder- und Lateralwand, des Septums, sämtlicher Papillarmuskeln sowie der Herzspitze (mindestens 30% des Myokards) zur Darstellung (Abb. 7). Bei ansonsten stenosefreien Koronararterien war dieser Befund gut vereinbar mit oben genannter Kompression des proximalen RIVA und des Ramus circumflexus (RCX) durch die Dissektion. Zusätzlich imponierte ein vollständig durch Blutkoagel okkludiertes linkes Bronchialsystem ohne mikro- oder makroskopisch fassbare Blutungsquelle. Der eingenähte Graft zeigte suffiziente Nahtverhältnisse ohne Leckage, weshalb man von einer diffusen Sickerblutung bei Vollheparinisierung und Thrombopenie unter vaECLS ausging. Die Histologie der Lungen zeigte eine unspezifische diskrete Vaskulopathie mit prominenten Interlobärsepten und eine Arterialisierung von Venolen sowie eine minimale Pulmonalarteriosklerose. Die vasoreaktive pulmonale arterielle Hypertonie (PAH) ist eine seltene Untergruppe der IPAH, die sehr gut auf die Therapie mit CCB reagiert. Die Histologie der vasoreaktiven PAH ist in der Literatur nicht beschrieben.
Abbildung 7: Vierkammerschnitt des Herzens mit prominenter Einblutung des vorderen Papillarmuskels sowie dunkel-schattiertes Myokard im Bereich der Vorderwand und des Septums (histologisch zeigte sich dort ein subakuter Myokardinfarkt). Suffiziente Nahtverhältnisse eines eingenähten Pulmonalarterien-Grafts.
Diskussion
Die akute Dissektion der Pulmonalarterie ist eine seltene und meist letale Komplikation bei Patienten mit pulmonaler Hypertonie. Der Grossteil der Dissektionen wird erst post-mortem festgestellt und vor allem bei Patienten mit Aneurysmata der Pulmonalarterie diagnostiziert [1]. Die Mehrheit dieser Patienten hat einen unkorrigierten angeborenen Herzfehler mit sekundärer pulmonaler Hypertonie. Eine IPAH, wie bei unserer Patientin, ist eine seltene Ursache einer Dissektion, die nur in seltenen Fällen in der Literatur beschrieben wurde [1–3].
Dissektionen der Pulmonalarterie ohne pulmonale Hypertonie wurden bei Bindegewebsstörungen oder lokalen vaskulitischen Veränderungen beschrieben, zum Beispiel bei Patienten mit Morbus Behcet, Marfan-Syndrom oder Syphilis. Diese stellen insgesamt jedoch eine Rarität dar [4].
Entsprechend dem Laplace-Gesetz entsteht eine Dissektion am Ort der grössten Wandspannung. Durch die Degeneration der elastischen Fasern der Pulmonalarterie entsteht eine arterielle Gefässwandfragilität, die wiederum zu Rissen in der Intima prädisponiert [5]. Ob eine transiente Erhöhung des pulmonalen Drucks durch Belastung oder Husten bei einem respiratorischen Infekt «Trigger» für eine Dissektion der Pulmonalarterie sein können, bleibt unklar [1].
Die Symptome einer Dissektion der Pulmonalarterie sind unspezifisch, am häufigsten werden eine akute Dyspnoe und/oder Thoraxschmerzen beschrieben [5]. Diese Differentialdiagnose und eine entsprechende Diagnostik sollte bei allen Patienten mit pulmonaler Hypertonie und Dilatation der Pulmonalarterien in Betracht gezogen werden.
Mittels Echokardiographie kann bei gutem Schallfenster ein «dissection flap» identifiziert werden, bei unklarem Befund ist eine Computertomographie die Untersuchungsmethode der Wahl. Die Prognose einer akuten Pulmonalarterien-Dissektion ist sehr schlecht, und die meisten Patienten sterben an einem kardiogenen Schock infolge einer Perikardtamponade oder eines Herzinfarkts, an unkontrollierbarer pulmonaler Blutung oder an Multiorganversagen.
In einzelnen Fällen gelang eine chirurgische Sanierung der Dissektion der Pulmonalarterie [3]. In einem Fall wurde eine notfallmässige Herz-Lungen-Transplantation durchgeführt [6]. Sehr wenige Patienten mit IPAH können erfolgreich konservativ behandelt werden [5, 7]. Eine Kompression der Koronararterien, wie in unserem Fallbericht, ist eine bekannte, ebenfalls meist fatale Komplikation.
Bei Patienten mit IPAH werden häufig Aneurysmata der Pulmonalarterien beschrieben. In einer Kohorte von 60 Patienten mit IPAH in Südkorea wurde eine Dilatation des Truncus pulmonalis von mehr als 4 cm bei 20% der Patienten detektiert. Bei einem Drittel dieser Patienten konnten mechanische Komplikationen infolge der Dilatation des Truncus pulmonalis festgestellt werden: Kompression des linken Hauptstammes, Kompression der Luftwege, Thrombose des Truncus pulmonalis oder Kompression des Nervus recurrens. Dies erklärt das Auftreten der Symptome Angina, Heiserkeit oder Asthma [8]. Einige Patienten mit IPAH und Kompression des Hauptstammes bei Aneurysma der Pulmonalarterie von mehr als 4 cm haben von einer perkutanen Revaskularisation profitiert [9]. In einer Studie mit Patienten mit kongenitalen Herzfehlern wiesen 5,5% ein Pulmonalarterienaneurysma von mehr als 4 cm mit ähnlichen mechanischen Komplikationen auf [10].
Bei Patienten mit IPAH steht die medikamentöse Therapie im Vordergrund, es existieren keine Richtlinien oder Empfehlungen bezüglich der prophylaktischen Behandlung eines Pulmonalarterienaneurysmas. Nach unserem Wissen existiert nur ein einziger Fallbericht in der Literatur, bei dem ein Patient mit vasoreaktiver IPAH an einem Pulmonalarterienaneurysma operiert wurde, trotz Normalisierung der Hämodynamik unter Therapie. Das Aneurysma war 9 cm gross und hatte zu einer externen Kompression der linken Koronararterie und zu einer schweren Pulmonalisinsuffizienz geführt [11]. Patienten mit therapierefraktärer IPAH und grossen Pulmonalarterienaneurysmata wurden lungentransplantiert [12], was die ultimative Therapie der IPAH darstellt.
Bei Patienten mit kongenitalen Herzfehlern und assoziierten Pulmonalarterienaneurysmata liegen mehr Daten zur Behandlung vor. In einem Review der Mayo Clinic (USA) wurden im Zeitraum von 1977 bis 2002 insgesamt 51 Patienten operiert. Basierend auf dieser Arbeit empfehlen die Autoren eine operative Sanierung bei Aneurysmata über 6 cm Durchmesser oder bei symptomatischen Aneurysmen sowie rechtsventrikulärer Dysfunktion [13, 14]. Endovaskuläre Interventionen mit Coil-Embolisation können auch in Betracht gezogen werden, vor allem bei Patienten mit hohem perioperativen Risiko [15].
Die spezifische Behandlung der pulmonalen Hypertonie wie in unserem Fall reicht je nach Therapieansprechen und Grösse des Aneurysmas bisweilen nicht aus, um diese seltene Komplikation zu vermeiden. Unsere Patientin war trotz ihrer Krankheit über Jahrzehnte im Alltag wenig eingeschränkt und hatte bis zu ihrem Tod eine sehr gute Lebensqualität und körperliche Belastbarkeit.
Das Wichtigste für die Praxis
• Die akute Dissektion der Pulmonalarterie ist eine seltene, jedoch hochletale Komplikation bei Patienten mit Aneurysmata der Pulmonalarterie.
• Aneurysmata der Pulmonalarterie treten vorwiegend bei Patienten mit kongenitalen Herzfehlern und bei idiopathischer pulmonal-arterieller Hypertonie (IPAH) auf.
• Die Differentialdiagnose einer Dissektion der Pulmonalarterie sollte bei allen Patienten mit pulmonaler Hypertonie und akuter Dyspnoe, Thoraxschmerzen oder hämodynamischer Instabilität in Betracht gezogen werden.
• Die Computertomographie ist die Bildgebung der Wahl. Bei gutem Schallfenster kann eine Pulmonalarteriendissektion mit Dissektionsflap auch mittels Echokardiographie diagnostiziert werden.
• Die einzige therapeutische Option der Dissektion ist die chirurgische Sanierung. Diese ist insbesondere aufgrund des speziellen Patientenkollektivs (IPAH) mit einer hohen Mortalität assoziiert.
Disclosure statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Korrespondenz
Dr. med. Charlotte Berlier UniversitätsSpital Zürich Klinik für Pneumologie Rämistrasse 100 CH-8091 Zürich charlotte.berlier[at]usz.ch
Literatur
1 Degano B, et al. Fatal dissection of the pulmonary artery in pulmonary arterial hypertension. Eur Respir Rev. 2009;18(113):181–5.
2 Correa Rde A, et al. Pulmonary hypertension and pulmonary artery dissection. J Bras Pneumol. 2013;39(2):238–41.
3 Senbaklavaci O, et al. Rupture and dissection in pulmonary artery aneurysms: incidence, cause, and treatment – review and case report. J Thorac Cardiovasc Surg. 2001;121(5):1006–8.
4 Inayama Y, Nakatani Y, Kitamura H. Pulmonary artery dissection in patients without underlying pulmonary hypertension. Histopathology. 2001;38(5):435–42.
5 Khattar RS, Fox DJ, Alty JE, Arora A. Pulmonary artery dissection: an emerging cardiovascular complication in surviving patients with chronic pulmonary hypertension. Heart. 2005;91:142–5.
6 Wuyts WA, et al. Extensive dissection of the pulmonary artery treated with combined heart-lung transplantation. J Thorac Cardiovasc Surg. 2006;132(1):205–6.
7 Steurer J, et al., Dissecting aneurysm of the pulmonary artery with pulmonary hypertension. Am Rev Respir Dis. 1990;142(5):1219–21.
8 Lee SE, et al. Detection of mechanical complications related to the potential risk of sudden cardiac death in patients with pulmonary arterial hypertension by computed tomography. Int J Cardiol. 2017;243:460–5.
9 Galie N, et al. Left Main Coronary Artery Compression in Patients With Pulmonary Arterial Hypertension and Angina. J Am Coll Cardiol. 2017;69(23):2808–17.
10 Gallego P, et al. Prevalence and prognostic significance of pulmonary artery aneurysms in adults with congenital heart disease. Int J Cardiol. 2018;270:120–5.
11 Araujo I, et al. Giant pulmonary artery aneurysm in a patient with vasoreactive pulmonary hypertension: a case report. BMC Cardiovasc Disord. 2011;11:64.
12 Wekerle, T, et al. Lung transplantation for primary pulmonary hypertension and giant pulmonary artery aneurysm. Ann Thorac Surg. 1998;65(3):825–7.
13 Deb SJ, Zehr KJ, Shields RC. Idiopathic pulmonary artery aneurysm. Ann Thorac Surg. 2005;80(4):1500–2.
14 Hou R, et al. Surgical treatment of pulmonary artery aneurysm: an institutional experience and literature review. Interact Cardiovasc Thorac Surg. 2016;23(3):438–42.
15 Park HS, et al. Pulmonary artery aneurysms: diagnosis and endovascular therapy. Cardiovasc Diagn Ther. 2018;8(3):350–61.