Ein 67-jähriger Patient stellt sich wegen einer Parese des linken Beins, eines Harnverhalts, einer zweitätigen Episode mit Singultus und einer mehrstündigen Episode mit verschwommenem Sehen auf der Notfallstation vor.
Hintergrund
Wir berichten über einen Fall von Neuromyelitis optica (NMO), eine seltene Erkrankung des Zentralnervensystems (ZNS). Sie war die Ursache einer Hyponatriämie mit SIADH-Konstellation (Syndrom der inadäquaten ADH-Sekretion), ausgelöst durch die entzündliche Mitbeteiligung des Hypothalamus.
Fallbericht
Anamnese
Der 67-jährige Patient hatte sich aufgrund einer neu aufgetretenen Parese des linken Beins und eines Harnverhalts auf der Notfallstation vorgestellt. Bis auf eine arterielle Hypertonie bestanden keine Vorerkrankungen. In der Systemanamnese berichtete er über eine zweitägige Episode mit Singultus und eine mehrstündige Episode mit verschwommenem Sehen in den letzten Tagen. Bereits zehn Tage zuvor war der Patient aufgrund einer ausgeprägten körperlichen Schwäche mit Inappetenz und depressiver Verstimmung hospitalisiert gewesen. Die damalige Symptomatik wurde auf eine mittelschwere hypotone Hyponatriämie, passend zu einem SIADH, zurückgeführt. Die Hyponatriämie wurde ätiologisch als Nebenwirkung der antihypertensiven Therapie mit einem ACE-Hemmer und im Rahmen eines vermehrten Alkoholkonsums interpretiert. Sie normalisierte sich während des Spitalaufenthalts unter Trinkmengenrestriktion und unter vorübergehend pausierter antihypertensiver Therapie. Damals bereits beklagte brennende Missempfindungen im Bereich des rechten Oberschenkels und der linken Schulter wurden am ehesten auf die Elektrolytverschiebung zurückgeführt. Differentialdiagnostisch war auch an eine aethyltoxisch bedingte Ätiologie gedacht worden. Über anderweitige Symptome einer primär zerebralen Erkrankung mit Hypothalamusbeteiligung wie Störungen des Hungergefühls oder des Schlaf-Wach-Rhythmus klagte der Patient nicht.
Status
Beim Eintritt in unser Spital präsentierte sich ein allseits orientierter, afebriler, euvolämer Patient mit unauffälligem kardiopulmonalem und abdominalem Status. Die klinisch-neurologische Untersuchung war hinweisend auf eine Myelopathie mit Elementen eines Brown-Séquard-Syndroms. Es fand sich eine hochgradige Beinparese links mit erloschenen Muskeleigenreflexen links bei beidseits positivem Babinski-Zeichen. Ausserdem bestand eine dissoziierte Sensibilitätsstörung unterhalb Th4 im Sinne einer Hypästhesie sowie Hypalgesie rechtsseitig. Der Sphinktertonus war abgeschwächt, der freie Stand nur mit viel Unterstützung möglich. Der Hirnnervenstatus war unauffällig.
Diagnostik
Laboranalytisch zeigte sich bei Eintritt erneut eine hypotone Hyponatriämie, bei hypertonem Urin lag erneut eine SIADH-Konstellation vor. Das restliche Labor war unauffällig (Tab. 1). Eine Hypothyreose als Ursache der euvolämen Hyponatriämie konnte bei normwertigem TSH ausgeschlossen werden, auf eine Nüchtern-Kortisonbestimmung zum Ausschluss einer Nebennierenrindeninsuffizienz wurde leider verzichtet.
Tabelle 1: Laborwerte bei der initialen Arztvorstellung.
Serum
Referenzwerte
Natrium
125 mmol/l
136–145 mmol/l
Kalium
4,3 mmol/l
3,5–5,1 mmol/l
Kreatinin
70 μmol/l
64–111 μmol/l
Harnstoff
3,8 mmol/l
3,0–9,2 mmol/l
Osmolarität
265 mosmol/kg
280–300 mosmol/kg
TSH
0,62 mU/l
0,35–4,94 mU/l
Urin
Natrium
69 mmol/l
Kalium
15 mmol/l
Osmolarität
277 mosmol/kg
TSH: Thyreoidea stimulierendes Hormon
Mittels einer thorakalen Computertomographie war bereits während der vorgängigen Hospitalisation ein paraneoplastisches SIADH aufgrund eines Bronchialkarzinoms oder einer sonstigen pulmonalen Erkrankung ausgeschlossen worden. Weder klinisch noch anamnestisch ergab sich der Verdacht einer anderweitigen malignen Erkrankung. Eine medikamentöse SIADH-Ätiologie konnte anamnestisch ausgeschlossen werden, für seltenere Ursachen des SIADH konnten keine Hinweise gefunden werden. Schliesslich rückte die Verdachtsdiagnose einer primär neurologischen Erkrankung in den Vordergrund. Wir veranlassten eine Magnetresonanztomographie (MRT) der Wirbelsäule und des Cerebrums, dabei stellten sich multiple intramedulläre langstreckige T2-Läsionen mit inhomogener Kontrastmittelaufnahme im Myelon dar (Abb. 1). Zerebral zeigten sich ebenfalls multiple supratentorielle entzündliche Läsionen, insbesondere im Marklager und periventrikulär sowie im proximalen Hypothalamus (Abb. 2). Eine Mitbeteiligung der Sehnerven konnte im MRT nicht dargestellt werden, bestätigte sich anschliessend jedoch aufgrund der beidseitigen Leitungsverzögerung in der Messung der visuell-evozierten Potentiale.
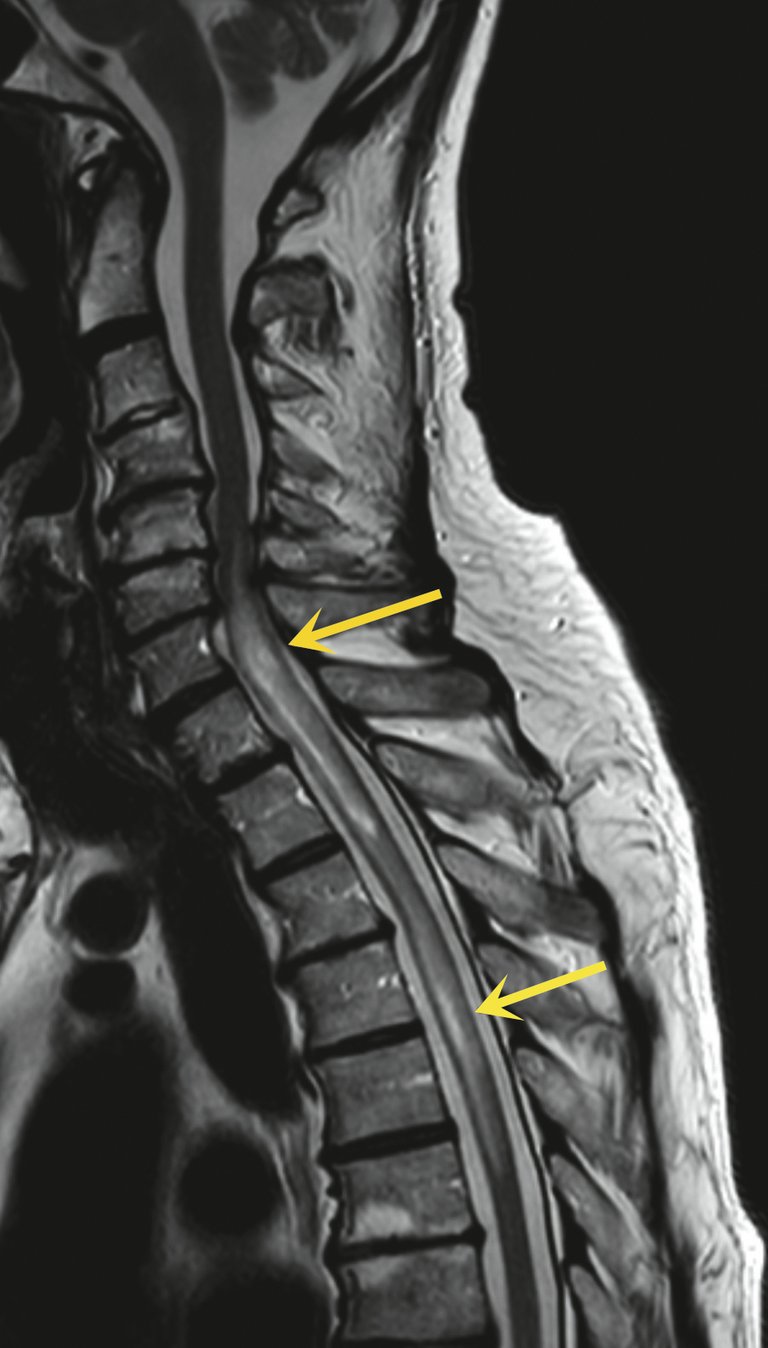
Abbildung 1: Sagittale T2-«turbo spin echo» (TSE)-Sequenz mit zwei langstreckigen intramedullären hyperintensen Arealen (gelbe Pfeile) mit deutlichem perifokalem Ödem im unteren Zervikalmark und im oberen Thorakalmark.
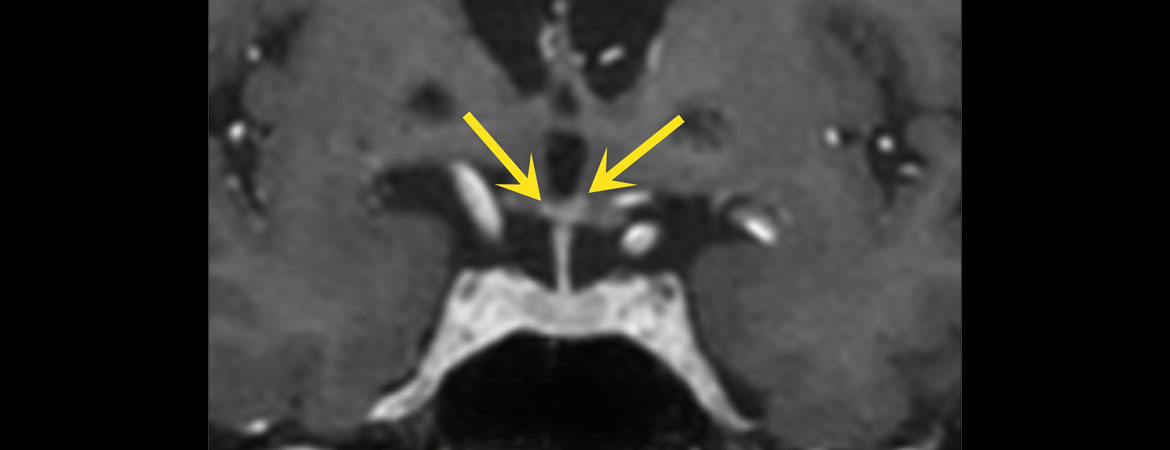
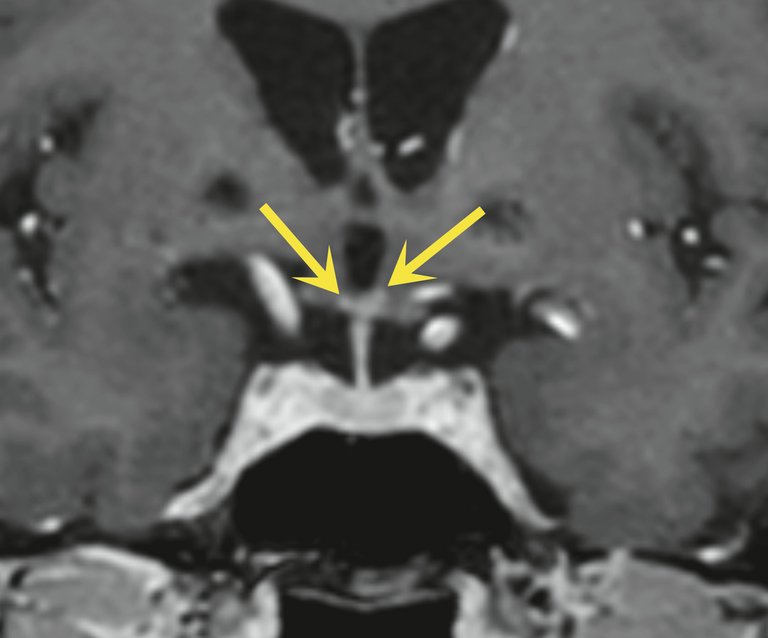
Abbildung 2: Koronare T1 m-Dixon-«turbo field echo» (TFE)Sequenz nach Kontrastmittelgabe mit bilateralen kontrastmittelanreichernden entzündlichen kleinen punktförmigen Anreicherungen im Hypothalamus (gelbe Pfeile).
Bei zentralen Demyelinisierungen und Mitbeteiligung der Sehnerven dachten wir an eine Multiple Sklerose (MS). Dazu waren jedoch die langstreckigen Demyelinisierungen im MRT nicht passend, ebenfalls waren oligoklonale Banden im Liquor nur in niederer Konzentration nachweisbar. Da sich in der Lumbalpunktion eine lymphozytäre Pleozytose von 50 Zellen/µl und eine deutliche Blut-Hirn-Schrankenstörung gezeigt hatte, wäre differentialdiagnostisch auch eine infektiologische Ursache denkbar gewesen. Bei normwertiger Glukose und normwertigem Lactat in der Lumbalpunktion, negativer bakterieller Liquorkultur und negativen Serologien (Lues, Borrelien, HIV, HSV, VZV und HHV-6) ergaben sich hierfür jedoch keine Anhaltspunkte. Ebenfalls erschien eine zerebrale Vaskulitis bei negativem Vaskulitis-Screening mittels Autoantikörpern (antinukleäre Antikörper, Anti-dsDNA-Antikörper, Anti-SSA (Ro)/-SSB (La)-Antikörper, Komplement-C3/C4 negativ) unwahrscheinlich.
Schliesslich stellten wir aufgrund der langstreckigen Myelitis und der bilateralen Opticusneuropathie die Diagnose einer Neuromyelitis optica. Passend dazu waren die Aquaporin-4-Antikörper (AQP4-AK) im Serum hoch positiv (1:320, Normbereich <1:10).
Therapie und Verlauf
Auf die initiale Steroidstosstherapie mit 1 g Methylprednisolon täglich während sieben Tagen und die nachfolgende perorale Steroid-Erhaltungstherapie zeigte sich keine neurologische Besserung, sodass wir auf eine Plasmapherese eskalierten. Leider trat als Nebenwirkung eine Gerinnungsstörung mit ausgedehnter anämisierender Hämatombildung am paretischen linken Bein auf, sodass wir die Plasmapherese nach fünf Zyklen frühzeitig beenden mussten. Bis dahin hatte sich eine leichte Verbesserung der Kraft im linken Bein gezeigt, wobei der Patient weiterhin rollstuhlmobil war. Bezüglich der initialen mittelschweren Hyponatriämie wurde bei Spitaleintritt kurzzeitig eine hyperosmolare Natriumchlorid-Lösung infundiert, bei Natriumanstieg in den tiefnormalen Bereich jedoch bereits nach wenigen Stunden wieder gestoppt. In der Folge blieben das Serumnatrium und die Serumosmolarität im tiefnormalen Bereich.
Knapp vier Wochen nach Spitaleintritt verabreichten wir erstmalig 1 g Rituximab zur Rezidivprophylaxe. Schliesslich war der Patient nach viermonatiger Neurorehabilitation ohne Hilfsmittel mobil, residuell bestand noch eine leichte linksbetonte Ataxie. Die Rituximabtherapie wird in sechsmonatigen Abständen während den nächsten zwei Jahren weitergeführt werden.
Diskussion
Bei der Neuromyelitis optica handelt es sich um eine entzündliche Autoimmunerkrankung des Zentralnervensystems mit typischem Befall der Sehnerven und des Rückenmarks. Klinisch manifestiert sich die NMO mit Visusstörungen, Paresen, Sensibilitätsstörungen und Blasendysfunktion. Bei Befall des Hirnstamms und insbesondere der Area postrema kann es zu Nausea, Schluckauf und Ateminsuffizienz kommen [1].
Die Mehrheit der Patienten erleidet einen progredienten Krankheitsverlauf mit wiederholten Schüben, wobei seltener auch monophasische Verläufe beschrieben sind. Insgesamt betreffen Erkrankungen aus dem NMO-Spektrum durchschnittlich 2/100 000 Personen, wobei Frauen häufiger als Männer und Nicht-Weisse häufiger als Weisse betroffen sind.
Die Erkrankung wurde ursprünglich 1894 von Eugene Devic als opticospinales Syndrom mit simultaner optischer Neuritis und transverser Myelitis beschrieben («Devic’s disease»). Die Abgrenzung insbesondere zur Multiplen Sklerose war lange unscharf. Erst mit Entwicklung von bildgebenden Verfahren und Liquoranalysen traten zunehmend Unterschiede zutage. So können bei der NMO im Gegensatz zur MS üblicherweise keine oligoklonalen Banden im Liquor nachgewiesen werden, das Schädel-MRT ist bei der NMO häufiger unauffällig, und die Rückenmarks-Demyelinisierungen sind bei der NMO langstreckiger als bei der MS. Treten bei der NMO doch zerebrale Demyelinisierungen auf, so sind sie typischerweise entlang des 3. und 4. Seitenventrikels lokalisiert. Erst die Entdeckung des hochspezifischen Serum-Autoantikörpers gegen den Aquaporin-4-Kanal (AQP4-IgG) im Jahre 2004 festigte die NMO weiter als eigene Entität. Jedoch sind etwa 30% aller NMO Patienten AQP4-IgG-negativ, wobei wiederum bei ungefähr einem Viertel ein Antikörper gegen oligodendrogliales Myelin (Anti-MOG-IgG) nachgewiesen werden kann [2]. Im Jahr 2007 wurde der Begriff der «NMO spectrum disorder» (NMOSD) geprägt, der nebst der klassischen NMO auch unvollständige Formen umfasst. Im Jahr 2015 wurden die heute gültigen Kriterien zur Diagnosestellung der NMOSD etabliert [3].
Die genaue Entstehung der Erkrankung ist unklar. Dem AQP4-Antikörper selber scheint eine zentrale Rolle in der Pathogenese zuzukommen, da er durch Bindung an astrozytäre Kanäle im ZNS direkt zur Entzündung, Demyelinisierung und Komplement-vermittelten Lyse der Zielzelle beiträgt.
Akute Schübe werden mit Steroidstössen behandelt. Bei fehlendem Ansprechen kann eine Plasmapherese erwogen werden. Zur Rückfallprophylaxe wird eine Immunsuppression für mindestens fünf Jahre empfohlen. Zumeist werden Steroide mit Azathioprin, Mycophenolat-Mofetil oder Rituximab kombiniert, wobei kein Direktvergleich in prospektiven Studien existiert. Interessanterweise sind klassische immunmodulatorische MS-Therapien wirkungslos beziehungsweise potentiell schädlich. Die Prognose der NMO ist schlechter als bei der Multiplen Sklerose, die Schübe sind häufig schwerer und die neurologische Erholung schlechter.
Hyponatriämien betreffen laut Literatur etwa 15% aller NMOSD-Patienten, bei den meisten besteht wie bei unserem Patienten ein SIADH [4]. Mögliche Mechanismen für das SIADH bei NMOSD sind immunologische Affektionen des AQP4-reichen Hypothalamus mit Austritt von ADH oder des zirkumventrikulären Organs, das ebenfalls an der Regulation des Salzhaushaltes beteiligt ist. Da die NMOSD im Gegensatz zur MS eine Prädilektion für einen hypothalamischen Befall aufweisen, tritt auch das SIADH häufiger auf als bei der MS. Eine viel seltenere Ursache einer Hyponatriämie bei vielen zerebral neurologischen Erkrankungen ist das zerebrale Salzverlust-Syndrom. Dieses unterscheidet sich laboranalytisch kaum vom SIADH, geht jedoch klinisch mit einer Dehydratation einher aufgrund einer Polyurie bei übermässiger renaler Natriumausscheidung.
Das Wichtigste für die Praxis
• Bei der Differentialdiagnose von Hyponatriämien, insbesondere bei SIADH-Konstellation, sollte auch an neurologische Erkrankungen gedacht werden.
• Bei Neuritis optica und langstreckiger Myelitis muss an eine Neuromyelitis optica (NMO) gedacht werden. AQP4-IgG-Antikörper sind hier hochspezifisch, jedoch gibt es auch NMO-Spektrumerkrankungen mit negativen AQP4-IgG-Antikörpern.
• Im akuten Schub wird die NMO mit Steroidstössen und in schwerwiegenden Fällen mit Plasmapherese behandelt. Zur Rezidivprophylaxe wird eine Immunsuppression durchgeführt.
• Die Abgrenzung zur Multiplen Sklerose ist therapeutisch wichtig, da die klassischen MS-Therapeutika eine NMO potentiell verschlechtern können.
Disclosure statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
1 Weinshenker BG, Wingerchuk DM. Neuromyelitis Spectrum Disorders. Mayo Clin Proc. 2017;92(4):663–79.
2 Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Luccinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–12.
3 Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177–89.
4 Jin S, Long Z, Wang W, Jiang B. Hyponatremia in neuromyelitis optica spectrum disorders: Literature review. Acta Neurol Scand. 2018;138(1):4–11.