Die homozygot-rezessiv vererbte Zystische Fibrose stellt die häufigste lebenslimitierende genetische Erkrankung in der kaukasischen Bevölkerung dar. Durch verbesserte diagnostische und therapeutische Strategien hat sich sowohl die Lebensqualität der Betroffenen als auch die mittlere Überlebensrate in den letzten zwei Dekaden dramatisch verbessert. Dieser Trend hält, bedingt durch Neuerungen wie die Einführung des Neugeborenen-Screenings und mutationsspezifische Therapien, weiter an, eröffnet aber auch neue Herausforderungen im klinischen Management.
Spektrum der genetischen und klinischen Ausprägungen
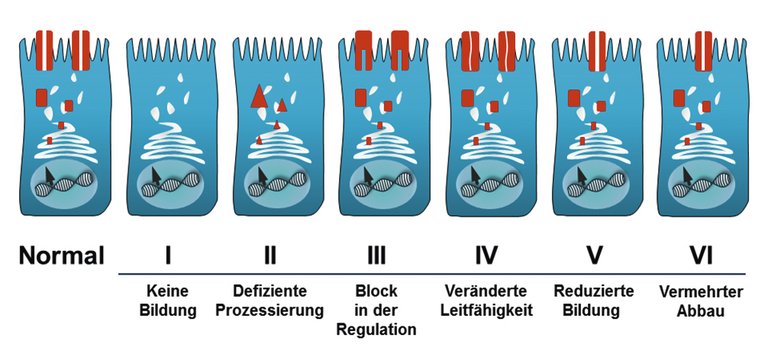
Ursache der Zystischen Fibrose (CF) ist eine Mutation in einem Gen auf dem langen Arm von Chromosom Nr. 7, welches für das «cystic fibrosis transmembrane conductance regulator»(CFTR)-Protein kodiert. Das CFTR-Protein bildet an der Oberflache der menschlichen Epithelzellen den Chlorid-Ionenkanal. Entsprechend ist bei Menschen mit einer homozygoten oder compound-heterozygoten Mutation im CFTR-Gen die Ausbildung oder Funktion des Chloridkanals essentiell gestört, wobei nur ca. 10% der bisher bekannten >2000 CFTR-Mutationen sicher krankheitsauslösend sind. Je nach Art der Störung in der CFTR-Synthese lassen sich CFTR-Mutationen in sechs Klassen mit unterschiedlicher Konsequenz für die Funktionalität des Chloridkanals einteilen (Abb. 1). Heterozygote Genträger sind gesund (Prävalenz in Mitteleuropa ca. 1:25). Neben dem Phänotyp der «klassischen» CF mit Multiorganmanifestation, typischen Symptomen und Komplikationen (Tab. 1) sind CFTR-Mutationen bekannt, die mit einer milderen Ausprägung der Erkrankung und einer Oligo- oder Monosymptomatik einhergehen. Die Symptome können hierbei teilweise auch erst jenseits der Kindheit auftreten und manifestieren sich oft als Pankreatitis, Sinusitis, Nasenpolypen, diffuse Bronchiektasie und/oder Infertilität des Mannes. Die Betroffenen weisen in der Regel keine Pankreasinsuffizienz und keine schwere Lungenerkrankung auf. Es ist deshalb wichtig, auch bei Erwachsenen mit suggestiven Symptomen, für welche keine anderweitige Ursache gefunden werden kann, an eine CFTR-Dysfunktion zu denken. Diese früher als «atypische» CF bezeichneten Phänotypen unterschiedlicher Manifestation und Ausprägung werden heute nach einem Konsensus der «European Cystic Fibrosis Society» (ECFS) als «CFTR-related disorders» (CFTR-RDs) bezeichnet [1]. Eine laufend aktualisierte Übersicht über die bekannten CF-auslösenden CFTR-Mutationen sowie deren Funktionalität und klinische Manifestationen findet sich auf der Website des CFTR2-Projekts (https://cftr2.org).
Abbildung 1: CFTR-Mutationsklassen mit resultierender Dysfunktion des Chloridkanals (mit freundlicher Genehmigung von Helge Hebestreit, Würzburg. Originalabbildung in: Speer-Gahr: Pädiatrie. VII. Zystische Fibrose. 2013. pp 499–505 [p501]. H. Hebestreit, A. Hebestreit. Springer-Verlag Berlin Heidelberg. Mit freundlicher Genehmigung von Springer Nature).
Tabelle 1: Klinische Symptome und Komplikationen der Zystischen Fibrose (CF) nach Alter. Die Prozentangaben in Klammern geben die Prävalenz im Jahr 2014 in der Schweiz an soweit Daten verfügbar sind (nach ECFSPR-Jahresbericht 2014).
Wie Kinder, zusätzlich: – Akute Pankreatitis – Hepathopathie (23,0%) – Portale Hypertension – CF-assoziierter Diabetes mellitus (CFRD; 23,7% bei ≥18 Jahren) – Osteoporose – CF-assoziierte Arthritis – Kongenitale bilaterale Absenz des Vas deferens (CBAVD)
ECFSPR = «European Cystic Fibrosis Society Patient Registry»
Diagnosestellung gestern und heute
«Wehe dem Kind, das salzig schmeckt, wenn man es auf die Stirn küsst: es ist verhext und muss bald sterben.» Diese überlieferte mittelalterliche Weisheit der Hebammen in Nordeuropa stellte bereits damals einen diagnostischen Zusammenhang zwischen einem hohen Salzgehalt des Schweisses und einer damals noch unbekannten letalen Erkrankung her. Heute basiert die Diagnosestellung der CF auf typischen klinischen Symptomen und der laborchemischen Bestätigung der zugrunde liegenden Dysfunktion des CFTR-Proteins. Die Bestimmung der Chloridkonzentration im Schweiss mittels quantitativer Pilocarpin-Iontophorese («Schweisstest») stellt den Goldstandard der Diagnose der CFTR-Dysfunktion dar [2]. Die Methode geht zurück auf die Beschreibung von Gibson und Cooke 1959, wobei die Schweissgewinnung über die Jahrzehnte technisch vereinfacht wurde und heute hierfür in der Schweiz im Wesentlichen das kommerziell erhältliche Macroduct®-System zum Einsatz kommt. In den letzten Jahren hat sich daneben mit der Messung der Schweissleitfähigkeit (Konduktivität) eine weitere Methode entwickelt und etabliert, wobei diese nach den aktuell gültigen internationalen Empfehlungen lediglich als Screeningmethode akzeptiert ist, während im Falle eines auffälligen Testergebnisses weiterhin eine konfirmatorische Bestimmung der Chloridkonzentration durchgeführt werden muss. Auch hier stehen zuverlässige kommerzielle Testsysteme zur Verfügung, wobei das Nanoduct®-System in der Schweiz weit verbreitet ist. Der zweistufige Schweisstest (Screening mittels Leitfähigkeit, Bestätigung mittels Chlorid nur im Fall einer erhöhten Leitfähigkeit) wird mittlerweile von vielen Labors durchgeführt, da bei einer normalen Leitfähigkeit eine klassische CF weitgehend ausgeschlossen werden kann. Andere Methoden wie die Bestimmung der Schweissosmolalität oder eine alleinige Bestimmung von Natrium oder Kalium ohne Chlorid sind heute obsolet. Bei der Interpretation der Resultate des Schweisstests gilt es zu beachten, dass die Referenzwerte der Leitfähigkeitsmessung über denen der Chloridbestimmung liegen, da die Leitfähigkeit neben Chlorid auch von anderen Ionen determiniert wird (Tab. 2). Das Schweisstestlabor sollte neben der verwendeten Methode auch den Referenzwertbereich und das gesammelte Schweissvolumen oder -gewicht beziehungsweise im Falle der Konduktivitätsmessung die Schweissrate mit den jeweiligen Cut-Off-Werten dokumentieren. Ein Testergebnis mit einer zu geringen Schweissmenge respektive -rate darf nicht verwendet werden und bedarf einer Wiederholung der Untersuchung.
Tabelle 2: Referenzwerte für verschiedene Schweisstest-Methoden.
Chloridkonzentration
Leitfähigkeit (NaCl-Äquivalente)
CF
≥60 mmol/l
≥80 mmol/l
Graubereich
30–59 mmol/l
50–79 mmol/l
Keine CF
<30 mmol/l
<50 mmol/l
Schweissvolumen
≥15 µl (Macroduct®)
≥3 µl (Nanoduct®)
Schweissrate
./.
≥1,0 g/m2/min
CF = Zystische Fibrose, ./. = nicht zutreffend
Neben einer suggestiven Symptomkonstellation können bei asymptomatischen Individuen auch das Vorhandensein einer CF bei einem Geschwister oder ein positives Neugeborenen-Screening eine Indikation für die Durchführung eines Schweisstests darstellen. Dieser sollte möglichst in einem qualifizierten CF-Zentrum durchgeführt werden. Im Falle eines positiven Schweisstest sollte der Patient zügig in das nächstgelegene CF-Zentrum zur weiteren Diagnostik überwiesen werden (Abb. 2). Für die Diagnosestellung wird in der Regel ein zweiter, konfirmatorischer Schweisstest durchgeführt. Bei einer Bestätigung der CF oder in unklaren Fällen (z.B. intermediärer Schweisstest, Schweisstest ohne verwertbares Resultat) schliesst sich neben einer Untersuchung der Pankreasfunktion (Pankreaselastase im Stuhl erniedrigt) eine Bestimmung der CFTR-Mutationen auf beiden Allelen an, was nicht nur eine Bedeutung für die Prognose des Krankheitsverlaufes und für eine genetische Beratung (z.B. weitere Familienplanung), sondern im Zeitalter der mutationsspezifischen Therapien immer öfter auch therapeutische Konsequenzen hat.
Abbildung 2: Flussdiagramm zur Diagnosestellung einer Zystischen Fibrose (CF) bei klinischem Verdacht (modifiziert und vereinfacht nach [2]).
CFTR = «cystic fibrosis transmembrane conductance regulator»
In einigen wenigen Fällen kann anhand der genannten Methoden die Frage, ob eine CF vorliegt, nicht eindeutig beantwortet werden. Hier können die Bestimmung der nasalen Potentialdifferenz (NPD) oder die intestinale Kurzschlussstrommessung (ICM) Klarheit über die CFTR-Funktion bringen. Diese Methoden sind jedoch nur in wenigen hochspezialisierten Zentren verfügbar.
Neugeborenen-Screening seit 2011
Am 1. Januar 2011 wurde in der Schweiz das Neugeborenen-Screening auf CF eingeführt [3]. Das Screening besteht aus einem zweistufigen Testverfahren im Rahmen der Fersenblutentnahme am 4. Lebenstag (immunreaktives Trypsinogen [IRT], im positiven Fall gefolgt von einer Mutationsanalyse, aktuell auf die in der Schweiz häufigsten 18 CFTR-Mutationen). Alle positiv gescreenten Kinder werden direkt vom zentralen Screening-Labor, das am Universtäts-Kinderspital Zürich angesiedelt ist, an das nächstgelegene CF-Zentrum zur weiteren diagnostischen Abklärung überwiesen. Hier erfolgt zunächst die Durchführung eines Schweisstest. Im Falle eines auffälligen Ergebnisses werden eine erweiterte Mutationsanalyse zur Detektion beider CFTR-Allele und eine Stuhlprobe auf Pankreaselastase durchgeführt. Aus den aktuellen Erkenntnissen des Neugeborenen-Screenings ergibt sich eine Inzidenz der klassischen CF in der Schweiz von 1:3631 Neugeborene (Neugeborenen-Screening Schweiz, Jahresbericht 2016, Download unter http://www.neoscreening.ch/de/jahresberichte.htm). Die Sensitivität des Screenings in der Schweiz für die Detektion einer klassischen CF beträgt 95%; das bedeutet, dass bis zu 5% falsch negative Testergebnisse vorkommen können. Deshalb schliesst ein unauffälliges Screening-Resultat eine CF nicht ganz sicher aus. Bei typischen klinischen Symptomen (Tab. 1) ist aus diesem Grund auch bei Patienten mit unauffälligem Neugeborenen-Screening an eine CF zu denken und eine weitere Diagnostik zu veranlassen. Alle Neugeborenen mit Mekoniumileus benötigen eine Abklärung mittels Schweisstest beziehungsweise Genetik, da diese Kinder bei Geburt ein normales IRT aufweisen können und somit mit dem Neugeborenen-Screening nicht erfasst werden.
Neben Kindern mit klassischer CF werden im Neugeborenen-Screening zwangsweise auch einige Kinder mit sehr milden Mutationen und grenzwertigem Schweisstest, das heisst vorhandener CFTR-Restfunktion, erfasst. Dieser Kinder haben keine CF, sondern werden als «CF screen positive, inconclusive diagnosis» (CFSPID) bezeichnet [4]. Einige von ihnen zeigen im Erwachsenenalter isolierte oder kombinierte Symptome eines CFTR-RD, sodass bei Vorliegen einer CFSPID von den Kinder- und Hausärzten die typischen Symptome einer CFTR-Dysfunktion überwacht werden sollten.
Erkenntnisse aus dem Patientenregister zur klinischen Versorgung
Für das Jahr 2015 werden erstmals alle CF-Zentren der Schweiz am «European Cystic Fibrosis Society Patient Registry» (ECFSPR) teilnehmen. Somit ist der Grundstein für künftige Benchmarking-Projekte mit dem Ziel der flächendeckenden Optimierung der Versorgungsqualität von Menschen mit CF in der Schweiz gelegt. Im Jahr 2014 wurden 789 Patienten mit CF aus der Schweiz ans ECFSPR gemeldet. Berechnet man die geschätzten Patientenzahlen aus den nicht teilnehmenden Zentren mit ein, darf man davon ausgehen, dass im Jahr 2014 etwa 950 Menschen mit CF in der Schweiz lebten, wobei zirka 50% dieser Menschen älter als 17 Jahre waren. Das Alter bei Diagnose lag im Jahr 2014 im Median bei nur sechs Monaten (Durchschnitt 2,7 Jahre), wobei bereits 15% aller lebenden Patienten im Rahmen des Neugeborenen-Screenings diagnostiziert wurden. Eine weitere Verschiebung Richtung Neugeborenenalter darf durch das Screening erwartet werden. Bei 99% aller Schweizer Patienten ist der genaue Genotyp bekannt. 86% aller Patienten in der Schweiz tragen die häufigste CFTR-Mutation delF508 auf mindestens einem Allel, 47% sind für diese Mutation homozygot. Für alle anderen Mutationen liegt die Häufigkeit bei unter 4%. Tabelle 3 zeigt wichtige klinische Eckdaten aus dem ECFSPR-Jahresbericht 2014. (abrufbar unter https://www.ecfs.eu/projects/ecfs-patient-registry/annual-reports).
Tabelle 3: Klinische Eckdaten der Zystischen Fibrose nach Altersgruppen in der Schweiz im Jahr 2014 (nach ECFSPR-Jahresbericht 2014). Die mikrobiologischen Angaben beziehen sich auf die Prävalenz einer chronischen pulmonalen Infektion.
<18 Jahre
≥18 Jahre
FEV1 v. Soll1,2
89,2% (77,4, 100,5)
62,1% (46,9, 81,8)
Gewicht (Z-Scores)
–0,4 (–1,1, 0,3)
–0,5 (–1,2, 0,1)
P. aeruginosa
12,8%
53,1%
S. aureus
55,2%
49,2%
NTM3)
0,8%
6,5%
1 «Global Lung Initiative equations»; alle Patienten ab 6 Jahren ohne Lungentransplantation 2 Median (25., 75. Perzentile) 3 NTM =Nontuberkulöse Mykobakterien ECFSPR = «European Cystic Fibrosis Society Patient Registry»
Behandlung im Zentrum ist essentiell
Wie in den meisten europäischen Ländern werden Menschen mit CF heute auch in der Schweiz in hochspezialisierten Zentren behandelt, die eine umfassende, interdisziplinäre Versorgung anbieten, hierfür entsprechendes spezifisch ausgebildetes Personal bereit stellen können und eine langjährige Expertise mit diesem komplexen Krankheitsbild aufweisen. Die Anforderungen an ein modernes CF-Zentrum sowie an präventive und therapeutische Massnahmen orientieren sich hierbei an internationalen Empfehlungen wie den «Standards of Care» der ECFS [5]. Die Patienten werden in der Regel in dreimonatlichen Abständen im Zentrum gesehen, wobei die durchgeführten Untersuchungen im Rahmen von standardisierten Programmen erfolgen, die im Detail je nach Erfahrung, Ressourcen und technischen Voraussetzungen von Zentrum zu Zentrum sowie abhängig vom Alter der Patienten leicht variieren können (Tab. 4). Ziel dieser Surveillance-Programme ist einerseits die frühzeitige Erkennung und das Monitoring des Verlaufes von Organmanifestationen und Komplikationen, andererseits aber auch die Therapieüberprüfung und -modifikation anhand des individuellen Krankheitsverlaufes. Spezifische Schulungen der Patienten und deren Angehörigen müssen in regelmässigen Abständen erfolgen, um den Herausforderungen eines komplexen Krankheitsmanagements im Alltag in Abhängigkeit von den unterschiedlichen Lebensphasen nachhaltig zu begegnen. Hierzu gehört neben krankheits- und therapiespezifischen medizinischen Themen auch die Beratung in sozialrechtlichen, finanziellen und psychologischen Fragen sowie hinsichtlich der Hygiene, der Verhaltensmassnahmen in Kindergarten, Schule und bei Reisen sowie eine Berufsberatung. Bei jungen Eltern mit noch nicht abgeschlossener Familienplanung und bei erwachsenen Patienten mit Kinderwunsch muss eine profunde genetische Aufklärung des Partners, in der Regel mit Unterstützung eines humangenetischen Zentrums, erfolgen. Bei einigen Patienten oder Familien kann eine interdisziplinäre stationäre Rehabilitation sinnvoll sein, um eine Verbesserung des Krankheitsmanagements oder eine Stabilisierung des Krankheitsverlaufes zu erreichen.
Tabelle 4: Standardisiertes Surveillance-Programm im CF-Zentrum (modifiziert nach [5]); hier am Beispiel Kinderspital Zürich.
Untersuchung
Details
Häufigkeit
Anleitung Physiotherapie
Inkl. Überprüfung Inhalationstechnik; im Zentrum oder in einer spezialisierten Praxis
Aufgrund der Errungenschaften der modernen Diagnostik und Therapie erreichen in den industrialisierten Ländern heute fast alle Patienten das Erwachsenenalter. Die Hälfte aller lebenden Menschen mit CF sind in der Schweiz älter als 17 Jahre. Das mittlere Überlebensalter lag 2012 in verschiedenen industrialisierten Ländern zwischen 40 und 50 Jahren [6]. Entsprechend wird die Transition von erwachsen gewordenen Patienten von dem pädiatrischen in ein adultes CF-Zentrum explizit von der ECFS gefordert [5] – in der Schweiz ist sie heute flächendeckend etabliert. Wenngleich die ECFS klare Empfehlungen zu Struktur und Ablauf abgegeben hat, variieren die Transitionsprogramme bezüglich Struktur und Inhalt in der Realität stark zwischen den Zentren. Daten aus dem amerikanischen CF-Register suggerieren eine Überlegenheit der Transition in ein Erwachsenenzentrum im Vergleich zu der häufig noch praktizierten Gewohnheit, die erwachsenen Patienten weiterhin am Kinderzentrum zu betreuen, zumindest in Bezug auf das pulmonale Outcome [7]. Darüber hinaus macht eine Transition auch aus klinischen Gesichtspunkten Sinn: Erwachsene CF-Patienten leiden mit zunehmendem Alter an internistischen Erkrankung wie Bluthochdruck oder Tumoren, die häufig von der CF unabhängig sind, diese aber komplizieren können. Hier ist internistisches Knowhow gefragt, was die Kompetenzen der Pädiatrie in der Regel übersteigt. Durch diese demographischen Veränderungen in der CF-Population müssen sich also auch die medizinischen Versorgungsstrukturen verändern: Interdisziplinäre internistische CF-Zentren müssen geschaffen werden; Fachdisziplinen wie zum Beispiel die Gynäkologie und Geburtshilfe, welchen die CF bisher unbekannt war, müssen sich mit den komplexen Anforderungen von Patienten mit CF befassen; Verschiebungen von Kosten weg von der Invalidenversicherung hin zu den Krankenkassen müssen antizipiert werden; Themen wie die berufliche Karriereplanung, der langfristige Erhalt der Erwerbsfähigkeit und die Familienplanung rücken in den Vordergrund und müssen adressiert werden. Die Planung des Erwachsenwerdens mit CF beginnt heute schon im Kindesalter: Während noch bis vor Kurzem die Prämisse galt, in erster Linie möglichst überhaupt das Erwachsenenalter zu erreichen, machen sich heute Familien mit CF-Kindern Gedanken über Themen wie Schul- und Berufsausbildung oder die Finanzierung einer eigenen Wohnung für das erwachsen gewordene Kind.
Neue und etablierte Therapien
Möglich gemacht haben diese demographische Entwicklung einerseits die etablierten Surveillance-Programme, andererseits aber insbesondere auch der Erfolg der modernen CF-spezifischen Therapie. Diese basiert im Wesentlichen auf drei Säulen: Enzymersatz- und Ernährungstherapie, tägliche inhalative und atemphysiotherapeutische Sekretolyse und eine aggressive Antibiotikastrategie. Eine Übersicht über die wichtigsten Therapien und Substanzen zeigt Tabelle 5. Auf die neueren Entwicklungen in den letzten Jahren wird im Folgenden kurz eingegangen.
Tabelle 5: Therapeutische Strategien zur Behandlung der Zystischen Fibrose (CF) inkl. verfügbarer Daten zur Häufigkeit in der Schweiz im Jahr 2014 (nach ECFSPR-Jahresbericht 2014).
Strategie
Therapieformen
Wichtigste Therapieziele
Häufigkeit in der Schweiz1
Ernährungstherapie
Pankreasenzyme
Normale Gewichts- und Grössenentwicklung
88,9%
Hochkalorische Ernährung
./.2
Fettlösliche Vitamine
Vermeidung von Hypovitaminose, Vitamin K-Mangel und Osteoporose
./.
Orale Kochsalzsubstitution
Prävention eines Salzverlustsyndroms
./.
Behandlung gastrointestinaler Komplikationen
Ursodeoxycholsäure
Behandlung der Cholestase; Prävention einer Hepathopathie
29,8%
Insulin
Behandlung des CFRD
23,7%3
Macrogol
Behandlung und Prävention des DIOS
./.
Sekretolyse
Respiratorische Physiotherapie (v.a. autogene Drainage und sekretolytische Hilfsmittel)
Verbesserung der mukoziliären Clearance; Vermeidung von Atelektasen, Bronchiektasen und pulmonalen Infektionen / Exazerbationen
./.
Inhalative hypertone Kochsalzlösung
60,9%
Inhalative rhDNAse
38,2%
Bronchodilatation
Beta-2-Sympathomimetika (kurz- und langwirksame)
Vorbehandlung vor hypertoner Kochsalzlösung; Behandlung der chronischen obstruktiven Ventilationsstörung
88,7%
Behandlung der pulmonalen Infektion
Systemische Antibiotika
Behandlung der pulmonalen Exazerbation; Prävention eines Lungenfunktionsverlustes
Inhalative Antibiotika
Eradikation von P. aeruginosa; Suppression bei chronischer Pseudomonas-Infektion zur Prävention von Exazerbationen
34,7%
Makrolide (v.a. Azithromycin bei chronischer Pseudomonas-Infektion)
Verbesserung der Lungenfunktion (vermutlich antiinflammatorische Wirkung)
28,3%
Systemische Antimykotika
Behandlung der ABPA
./.
Systemische Kortikosteroide
./.
Sinunasale Sekret-Clearance
Nasenspülung, Sinusinhalation
Prävention / Behandlung der Polyposis nasi
./.
Korrektur des Basisdefekts
CFTR-Korrektoren und -Potenziatoren
Verbesserung / Stabilisierung der Lungenerkrankung; Gewichtszunahme
<1%
1 in % aller CF-Patienten 2 nicht dokumentiert 3 bei ≥18 Jahren ECFSPR = «European Cystic Fibrosis Society Patient Registry», CFRD = CF-assoziierter Diabetes mellitus, DIOS = distale intestinale Obstruktion, ABPA = allergische bronchopulmonale Aspergillose
Die früher übliche sekretolytische Inhalationstherapie mit physiologischer Kochsalzlösung ist komplett durch höherprozentige Kochsalzlösungen zwischen 3 und 7% ersetzt worden, was über osmotische Prozesse zu einer wesentlich effizienteren Sekret-Clearance führt. Mannitol (Bronchitol®) hat als alternatives inhalatives Mukolytikum Eingang in das therapeutische Portfolio gefunden, wenngleich es in der Schweiz derzeit noch nicht zugelassen ist. Durch das Aufkommen der hypertonen Kochsalzlösung ist der Einsatz der über einen enzymatische Prozess ebenfalls sekretolytisch wirkenden, aber sehr viel teureren rhDNAse (Pulmozyme®) etwas zurückgegangen, obwohl in Studien der therapeutische Effekt von rhDNAse auf die Lungenfunktion der hypertonen Kochsalzlösung überlegen ist [8].
Durch das Älterwerden der Patienten spielt die chronische pulmonale Besiedlung mit einem oder mehreren Problemkeimen wie multiresistenten Pseudomonas-Stämmen, Burkholderia cepacia oder Mycobacterium abscessus eine immer gewichtigere Rolle. Während in der Schweiz die Prävalenz der chronischen pulmonalen Pseudomonas-Besiedlung bei Kindern und Jugendlichen nur noch bei 12,8% liegt, beträgt sie bei Erwachsenen 53,1% (Tab. 3). Bei jüngeren Patienten kann bei Infektion mit einem Problemkeim häufig noch mit konventionellen intravenösen oder inhalativen Antibiotika eine Eradikation erreicht werden. Insbesondere für Patienten mit einer chronischen Pseudomonas-Infektion ist die Entwicklung neuer und innovativer Antibiotika essentiell, um eine effektive Suppression der bakteriellen Aktivität zu erreichen und in der Folge pulmonalen Exazerbationen und einem Verlust an Lungenfunktion vorzubeugen. Hier kommen in erster Linie inhalative Strategien zum Einsatz, wobei die Anwendung alternierend ON/OFF für je vier Wochen oder als – teils kombinierte – Dauertherapie erfolgt. Neben etablierten Pseudomonas-wirksamen Substanzen wie Colistin und Tobramycin wurde vor wenigen Jahren Aztreonam (Cayston®) in der Schweiz zugelassen. Für Tobramycin steht seit Kurzem neben der Feuchtinhalation ein Trockenpulver zur Verfügung (TOBI® Podhaler®).
In jüngster Zeit machen sich neue therapeutische Strategien daran, den Verlauf und die Prognose der CF-Erkrankung potentiell zu revolutionieren. Anders als die in den 90er-Jahren starke Hoffnung erweckende Gentherapie, die es bisher zu keinem Durchbruch geschafft hat, zielen diese Therapien auf eine Modulation des CFTR-Kanals auf Proteinebene, wobei sich Korrektoren (verbesserte Verfügbarkeit von funktionsfähigem CFTR in der Zellmembran) von Potenziatoren (Verbesserung der Kanalöffnung und Funktionalität) unterscheiden lassen. Der Wirkmechanismus ist spezifisch für jeweils eine oder mehrere CFTR-Mutationsklassen, weshalb der Einsatz bei Patienten mutationsspezifisch erfolgt.
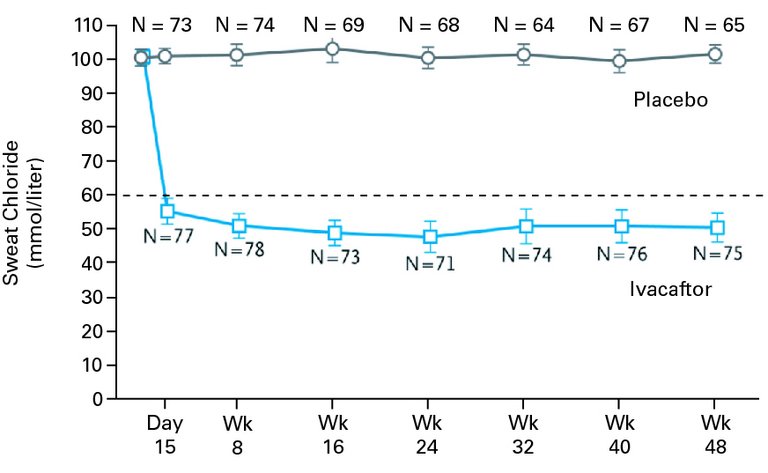
Mit Ivacaftor (Kalydeco®) ist seit 2015 der erste CFTR-Potenziator für bestimmte Klasse-III-Mutationen (sogenannte Gating-Mutationen) in der Schweiz zugelassen. Ivacaftor führt zu einer Öffnung des Chloridkanals und erhöht somit die Funktionalität des CFTR-Proteins. Klinische Studien zeigen unter Ivacaftor eine Verbesserung des Schweisstests um ca. 50 mmol/l, eine Stabilisierung des klinischen Zustandes sowie einen Zuwachs an FEV1 um etwa 10% ([9], Abb. 3). Dieser Effekt auf die Lungenfunktion liegt sehr deutlich über den konventionellen symptomatischen Therapieansätzen wie Sekretolytika und Antibiotika. Darüber hinaus wirkt sich Ivacaftor altersunabhängig positiv auf den Gewichtsverlauf aus [9, 10]. Die klinischen Erfahrungen seit Marktzulassung bestätigen die Studiendaten, wenngleich in der Schweiz aufgrund der dort niedrigen Frequenz von Gating-Mutationen nur sehr wenige Patienten von Ivacaftor profitieren können. Aufgrund der hohen Kosten von knapp 300 000 CHF pro Patient und Jahr ist die Abgabe mit einer Limitation versehen, welche die Wirksamkeit für jeden Patienten individuell überwacht.
Als zweite Substanz aus den CFTR-Modulatoren wurde Ivacaftor/Lumacaftor (Orkambi®) 2015 in den USA und der EU und 2016 in der Schweiz für CF-Patienten mit einer homozygoten delF508-Mutation zugelassen. Der Klasse-II-Korrektor Lumacaftor führt zu einer verbesserten Faltung des CFTR-Proteins, sodass weniger defektes CFTR in der Epithelzelle abgebaut wird und mehr Chloridkanäle an der Zelloberfläche zur Verfügung stehen. Ivacaftor soll für eine zusätzliche Optimierung der Kanalfunktionalität sorgen. Klinische Studien zeigen allerdings einen deutlich geringeren Effekt auf die Chloridkonzentration im Schweiss und die Lungenfunktion (ca. 3% FEV1-Verbesserung) im Vergleich zu dem Effekt von Ivacaftor bei den Gating-Mutationen. Allerdings verringert sich die Rate an pulmonalen Exazerbationen im Langzeitverlauf um bis zu 40%, was Orkambi® insbesondere für CF-Patienten mit instabilem pulmonalem Verlauf interessant macht [11]. Zudem scheint sich die Lungenfunktion mittelfristig zu stabilisieren, das heisst der krankheitsbedingt erwartete Lungenfunktionsabfall über die Zeit wird reduziert. Die bisherige klinische Erfahrung zeigt einerseits bei einigen Patienten mit fortgeschrittener Lungenerkrankung tatsächlich eine Reduktion der pulmonalen Exazerbationen und eine Zunahme des Gewichts unter Orkambi®, jedoch andererseits auch ein heterogenes Ansprechen der Patienten, sodass derzeit noch nicht ganz klar ist, welche Patienten am besten von der Therapie profitieren.
Weitere CFTR-Modulatoren sind derzeit in klinischen Phase-I-, -II- und -III-Studien in Erprobung. Hierzu gehört auch die Entwicklung einer weiteren Substanzklasse, welche die Wirkung der Korrektoren und Potenziatoren verstärken soll (sogenannte «amplifier»). Die Zukunft könnte in einer Kombination mehrerer Substanzen liegen, die individuell an den Patienten angepasst wird. Es besteht die Hoffnung, dass möglicherweise in den kommenden Jahren für eine wesentliche Anzahl an CF-Patienten wirksame mutationsspezifische Therapien zur Verfügung stehen werden.
Therapeutische Strategien zu Beginn und am Ende des Krankheitsverlaufes
Eine aus therapeutischer Sicht neue Herausforderung stellen Säuglinge und Kleinkinder dar, die aufgrund eines positiven Neugeborenen-Screenings diagnostiziert wurden. Ziel des Neugeborenen-Screenings ist es, Säuglinge mit CF und Pankreasinsuffizienz früh zu erfassen und noch im Neonatalalter die Behandlung mit einer Pankreasenzym-Ersatztherapie sowie fettlöslichen Vitaminen und einer Kochsalzsubstitution zu beginnen. Diese Kinder haben häufig in den ersten Lebensjahren keine oder nur sehr milde Atemwegssymptome. Daten unter anderem aus der australischen AREST-CF-Studie zeigen jedoch, dass strukturelle CF-typische Veränderungen im Lungen-Computertomogramm bereits den ersten beiden Lebensjahren nachweisbar sind, wenn die Mehrheit der Kinder noch asymptomatisch ist [12]. In der Schweiz wird im Rahmen der multizentrischen SCILD-Studie derzeit untersucht, welche frühen Einflussfaktoren wesentlich für den weiteren Krankheitsverlauf sind [13]. Folgerichtig werden auch bei respiratorisch unauffälligem Befund zeitgleich mit der Enzymersatztherapie eine sekretolytische Inhalationstherapie und eine Atemphysiotherapie eingeleitet und der gastrointestinale und pulmonale Krankheitsverlauf sowie die mikrobielle Besiedlung der Atemwege dreimonatlich überwacht. Ein laborchemisches Screening von Komplikationen und bildgebende Verfahren wie in Tabelle 4 dargestellt sind auch bereits in dieser Patientenpopulation in der Regel Bestandteil des klinischen Managements.
Auch im Angesicht der Verbesserung der Lebenserwartung und der Lebensqualität durch frühzeitige Diagnose, der standardisierten Surveillance und der verbesserten therapeutischen Strategien stellt für viele Menschen mit CF und terminaler Lungenerkrankung eine Lungentransplantation die letzte Option zur Verlängerung des Lebens dar. Da die Wartezeit auf ein neues Organ weiterhin Monate bis Jahre beträgt, ist die rechtzeitige Vorstellung im Transplantationszentrum für ein erstes Assessement und eine individuelle Beratung sinnvoll. Die durchschnittliche Wartezeit auf eine Lungentransplantation betrug 2015 nach Angaben von Swisstransplant 404 Tage (Median 202 Tage) (Swisstransplant Jahresbericht 2015, abrufbar unter https://www.swisstransplant.org/de/swisstransplant/publikationen/jahresberichte/). Im Jahre 2014 waren in der Schweiz 3,5% aller lebenden CF-Patienten lungentransplantiert. Die weltweite 5-Jahres-Überlebensrate von Kindern und Jugendlichen, die zwischen 1998 und 2014 lungentransplantiert wurden, liegt zwischen 50 und 55%, während sie bei Erwachsenen etwa 65% beträgt [14]. Für nähere Informationen zu Transplantation bei CF sei auf die publizierte Fachliteratur verwiesen.
Ausblick
Trotz erheblicher Verbesserungen im Bereich Vorsorge und Therapie ist der Verlauf der CF-Erkrankung im Einzelfall weiterhin sehr heterogen. Schwere Verläufe können auch heute noch bereits im Kindes- und Jugendlichenalter auftreten, was manchmal mit mangelnder Adherence, jedoch vermutlich häufiger mit bisher unbekannten krankheitsmodifizierenden Faktoren zusammenhängt. Die Detektion von Prädiktoren für den individuellen Krankheitsverlauf, wie beispielsweise inflammatorischen Markern, muss weiter vorangetrieben werden. Das Erwachsenenalter hingegen ist heutzutage häufig gekennzeichnet durch eine Konkurrenz zwischen einer zeitaufwendigen Therapie und einem möglichst uneingeschränktem Berufs- und Familienleben, was neben einer effizienten medizinischen Strategie auch ein anspruchsvolles Alltagsmanagement notwendig macht. Für beide Altersgruppen können neue mutationsspezifische Therapien einen wichtigen Beitrag leisten, indem sie den Krankheitsverlauf stabilisieren; allerdings ist der Zugang aller Betroffenen in Anbetracht der hohen Preise dieser Therapien künftig nicht sichergestellt. Ärzteschaft, Patientenorganisationen und Politik sind hier gefordert, einen gemeinsamen Weg zu finden, der keinem Patienten eine wirkungsvolle Therapie verwehrt.
Das Wichtigste für die Praxis
• Die Lebensqualität und das Überlebensalter von Menschen mit Zystischer Fibrose (CF) hat sich in den letzten Jahren verbessert. Ein optimiertes Management sowie neue effizientere Therapien haben für eine Stabilisierung der Krankheitsverläufe gesorgt.
• Potente sekretolytische Therapien, moderne inhalative Antibiotika und mutationsspezifische Therapieansätze, welche die CFTR-Funktion wiederherstellen oder verbessern, geben den Betroffenen heute eine neue Perspektive.
• Erkenntnisse aus dem europäischen CF-Register, an dem die Schweiz partizipiert, ermöglichen eine Verbesserung und Harmonisierung der Versorgungsqualität mittels Benchmarking.
• Der Anteil der Erwachsenen mit CF liegt mittlerweile bei rund 50%; den speziellen Anforderungen und Bedürfnissen dieser immer grösser und älter werdenden Patientengruppe muss vermehrt Rechnung getragen werden.
• Das 2011 eingeführte CF-Neugeborenen-Screening hat zu einer früheren Diagnosestellung und Therapieeinleitung geführt, was sich positiv auf den pulmonalen und gastrointestinalen Verlauf der Erkrankung – gerade während der Wachstumsphase – auswirken wird.
• Da der Anteil der falsch negativen Ergebnisse im CF-Neugeborenen-Screening bei bis zu 5% liegt, muss auch bei gescreenten Kindern bei suggestiver klinischer Symptomatik an eine CF gedacht und ein Schweisstest eingeleitet werden.
• Oligo- oder monosymtpomatische Phänotype können sich erst im Erwachsenenalter manifestieren; auch hier sollte bei Verdacht auf CFTR-Dysfunktion eine CF-Diagnostik durchgeführt werden.
• Patienten mit klinischen Verdacht auf CF, CFTR-Dysfunktion oder einem auffälligen Schweisstest sollen in jedem Fall an ein CF-Zentrum zur weiteren diagnostischen Abklärung und Therapie überwiesen werden.
Disclosure statement
Der Autor hat Beraterhonorare von Vertex, Novartis, Gilead und Vifor deklariert.
Korrespondenz
Dr. med. Andreas Jung Universitäts-Kinderspital Zürich Steinwiesstr. 75 CH-8032 Zürich andreas.jung[at]kispi.uzh.ch
Literatur
1 Bombieri C, Claustres M, De Boeck K, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10 Suppl 2:S86–S102.
2 De Boeck K, Wilschanski M, Castellani C, et al. Cystic fibrosis: terminology and diagnostic algorithms. Thorax. 2006;61:627–35.
3 Ruegg CS, Kuehni CE, Gallati S, et al. Neugeborenen-Screening auf Cystische Fibrose – Evaluation nach einem Jahr. Paediatrica. 2013;24:24–8.
4 Munck A, Mayell SJ, Winters V et al. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J Cyst Fibros. 2015; 14:706–13.
5 Kerem E, Conway S, Elborn S, Heijerman H. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros. 2005;4:7–26.
6 Sykes J, Stanojevic S, Goss CH, et al. A standardized approach to estimating survival statistics for population-based cystic fibrosis registry cohorts. J Clin Epidemiol. 2016;70:206–213.
7 Tuchman L, Schwartz M. Health outcomes associated with transition from pediatric to adult cystic fibrosis care. Pediatrics. 2013;132:847–53.
8 Suri R, Wallis C, Bush A, et al. A comparative study of hypertonic saline, daily and alternate-day rhDNase in children with cystic fibrosis. Health Technol Assessment. 2002;6:1–69.
9 Ramsey BW, Davies J, McElvaney N, Tullis E, Bell SC, Drevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and G551D mutation. N Engl J Med. 2011;365:1663–72.
10 Davies JC, Cunningham S, Harris WT, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4:107–15.
11 Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220–31.
12 Mott LS, Park J, Murray CP, et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax. 2012;67:509–16.
13 Mika M, Korten I, Qi W, Regamey N, Frey U, Casaulta C, SCILD study group. The nasal microbiota in infants with cystic fibrosis in the first year of life: a prospective cohort study. Lancet Respir Med. 2016;4:627–35.
14 Hayes D Jr, Glanville AR, McGiffin D, et al. Age-related survival disparity associated with lung transplantation in cystic fibrosis: An analysis of the registry of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2016;35:1108–15.