Die Systemsklerose ist eine zwar seltene, aber potentiell schwer verlaufende Konnektivitis, die oft mit einer beträchtlichen Morbidität und erhöhten Mortalität einhergeht. Eine Heilung ist nicht in Sicht, in vielen Fällen ist nur eine supportive Behandlung möglich. Das kann Unsicherheit und Angst bei Betroffenen und beim Arzt eine gewisse Hilflosigkeit verursachen. Dieser Artikel möchte dem Praktiker Tipps zur Diagnosestellung geben, therapeutische Vorschläge für die häufigsten Symptome darstellen und zum interdisziplinären Therapiemanagement anregen.
Einführung
Die Systemsklerose ist eine seltene Konnektivitis, die durch Vaskulopathie, Fibrose und Autoimmunität charakterisiert ist. Sie manifestiert sich häufiger bei Frauen als bei Männern (4:1), meistens im Alter von 35–65 Jahren. Alle Altersgruppen können aber davon betroffen sein [1].
Je nach Ausdehnung des Hautbefalls wird zwischen einer limitierten und einer diffusen Form unterschieden. Die Organmanifestationen können bei beiden Formen vorkommen, jedoch in unterschiedlicher Prävalenz. Sie können den Gastrointestinaltrakt, die Lunge, Niere, das muskuloskelettale System und das Herz betreffen. Der Terminus «CREST-Syndrom» sollte vermieden werden, da die CREST-Manifestationen der limitierten Form angehören. Zudem könnte dieser Terminus den Eindruck eines benignen Verlaufes erwecken, dabei können solche Patientinnen und Patienten nicht nur die unter dem Akronym «CREST» subsummierten Krankheitsmanifestationen (Calcinosis, Raynaud-Phänomen, Oesophageale Dysmotilität, Sklerodaktylie, Teleangiektasien), sondern auch weitere, schwerwiegendere Komplikationen wie Fingerulzerationen oder eine pulmonal-arterielle Hypertonie entwickeln.
Die Therapie richtet sich nach dem Organbefall. Es gibt zwar kein krankheitsmodifizierendes Medikament gegen Systemsklerose, in den letzten Jahren kam es jedoch zu erheblichen Fortschritten vor allem in der Behandlung der pulmonal-arteriellen Hypertonie. Dazu gibt es neue vielversprechende Wirkstoffe gegen die interstitielle Lungenerkrankung, welche Gegenstand laufender Studien sind.
Mittels Einschluss der Patienten ins EUSTAR («European Scleroderma Trials and Research Group»), eine europäische Kohorte von Patienten mit Systemsklerose, können wertvolle wissenschaftliche Informationen über diese seltene Erkrankung gewonnen werden.
Neue Klassifikationskriterien
Die neuen ACR/EULAR1-Klassifikationskriterien (2013) haben die älteren ARA2-Klassifikationskriterien (1980) bei besserer Sensitivität und Spezifität abgelöst (Tab. 1). Es darf jedoch nicht vergessen werden, dass diese Kriterien nicht der Diagnosestellung, sondern hauptsächlich Forschungszwecken dienen. Frühe Krankheitsformen können auch mit den neuen Kriterien «verpasst» werden. Hingegeben erlaubt ein ausgedehnter Hautbefall bereits die Diagnose einer Systemsklerose. Neu finden auch andere Krankheitsmanifestationen ihren Platz unter den neuen Klassifikationskriterien, sei es klinisch (Raynaud-Phänomen, Fingerschwellung) oder paraklinisch (spezifische Antikörper, pulmonal-arterielle Hypertonie oder pathologische Kapillarmikroskopie). Insbesondere das Raynaud-Phänomen, die Fingerschwellung und die positiven antinukleären Antikörper (ANA) können subtile Zeichen einer beginnenden Systemsklerose darstellen. Eine Zuweisung zur weiteren Abklärung ist schon in diesem Stadium indiziert, da eine frühe Diagnosestellung ein möglichst frühzeitiges Screening auf Organschäden erlaubt, bevor diese einer Therapie kaum oder nicht mehr zugänglich sind (Tab. 1).
Tabelle 1: Klassifikationskriterien.
ACR/EULAR-Klassifikationskriterien 2013 Eine Summe von ≥9 Punkten erlaubt die Diagnose einer Systemsklerose.
Kriterien
Unterkriterien
Punktzahl
Hautverdickung an den Fingern an beiden Händen. Befall über die Region proximal der Fingergrundgelenke hinaus (ausreichendes Kriterium).
−
9
Hautverdickung an den Fingern
Geschwollene Finger
2
Sklerodaktylie
4
Läsionen an den Fingerspitzen
Ulzera
2
Grübchenförmige Narben
3
Teleangiektasie
–
2
Abnorme Kapillaren im Nagelfalz
–
2
Pulmonal-arterielle Hypertonie oder interstitielle Lungenerkrankung (maximal 2 Punkte)
ACR/EULAR = American College of Rheumatology / European League Against Rheumatism
Klinische Manifestationen
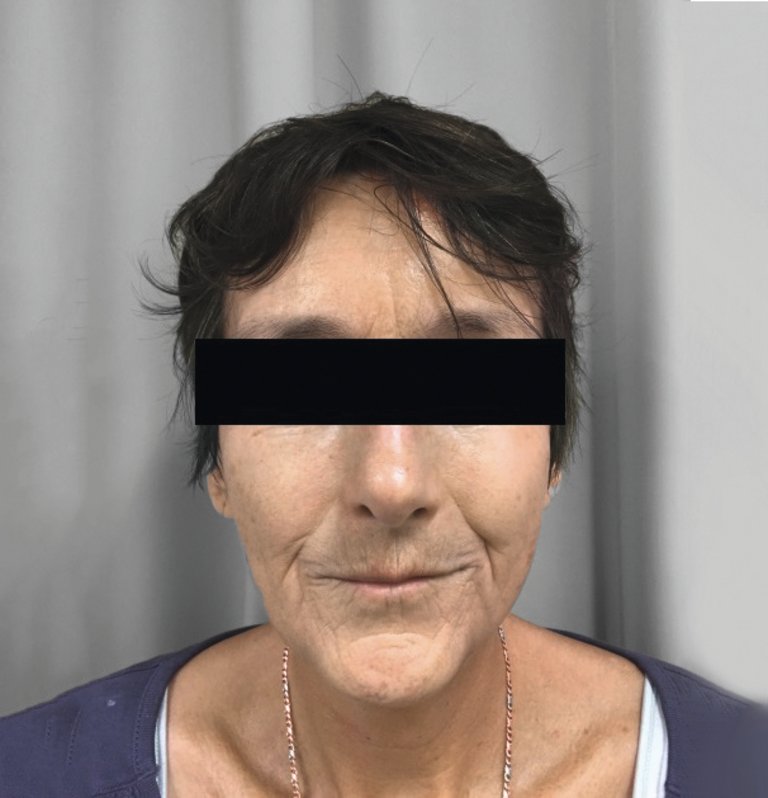
Die Hautveränderungen treten bei der limitierten Systemsklerose distal der Ellbogen respektive der Knie auf. Bei der diffusen Form sind diese auch proximal davon und am Rumpf vorhanden. Das Gesicht kann bei beiden Krankheitsformen mit dem typischen «Tabakbeutelmund» und Verlust der Mimik (Abb. 1) betroffen sein.
Abbildung 1: 55-jährige Patientin, seit 8 Jahren an limitierter Systemsklerose erkrankt, mit Tabakbeutelmund (die Publikation erfolgt mit dem Einverständnis der Patientin).
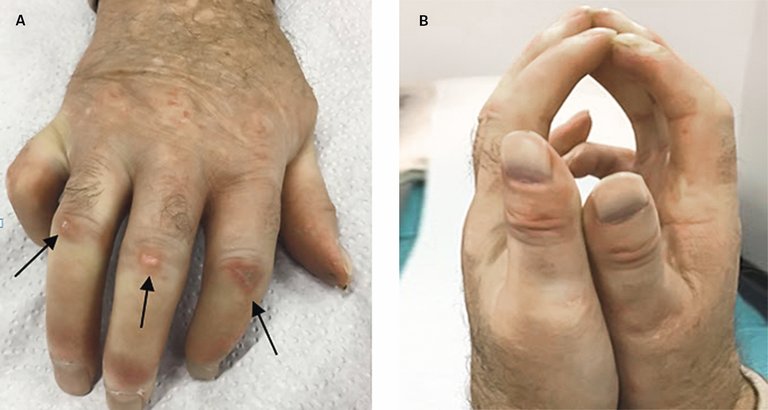
Initial gibt es meistens eine entzündliche Vermehrung der Kollagenfasern mit Ödembildung, später kommt es zur Fibrose mit Verhärtung der Haut. An den Fingern findet man oft Flexionskontrakturen mit sekundären Einbussen in der Greiffunktion (Abb. 2). Nicht allzu selten fehlt die Hautbeteiligung gänzlich. Man spricht dann von einer «Systemsklerose sine scleroderma».
Abbildung 2 A und B: Hautfibrose mit sekundären Flexionskontrakturen und Fingerulzerationen (Pfeile in A) bei einem Patienten mit diffuser Systemsklerose.
Das Raynaud-Phänomen (Abb. 3) ist die erste Krankheitsmanifestation, bei mehr als 85% der Patienten vorhanden, im Verlauf oft zunehmend und schliesslich zu Ischämie und bei bis zur Hälfte der Fälle zu akraler Nekrose (Fingerkuppenulzera) führend (Abb. 4). Bei der limitierten Form tritt das Raynaud-Phänomen oft lange Zeit vor anderen Krankheitsmanifestationen auf. Die Hautveränderungen entwickeln sich üblicherweise langsam. Als weiterer Ausdruck der Vaskulopathie kann sich Jahre später eine pulmonal-arterielle Hypertonie manifestieren. Bei der diffusen Form tritt das Raynaud-Phänomen oft kurz vor oder gleichzeitig mit den Hautveränderungen auf. Die Betroffenen entwickeln rasch im Verlauf viszerale Manifestationen wie die interstitielle Lungenerkrankung («interstitial lung disease» [ILD]) oder die renale Krise.
Abbildung 3: Raynaud-Phänomen. Bei dieser 33-jährigen Patientin fällt eine blasse Verfärbung des rechten Ringfingers (Pfeil) bei einer diffusen, leicht violetten Verfärbung der restlichen Finger und der Handflächen auf.
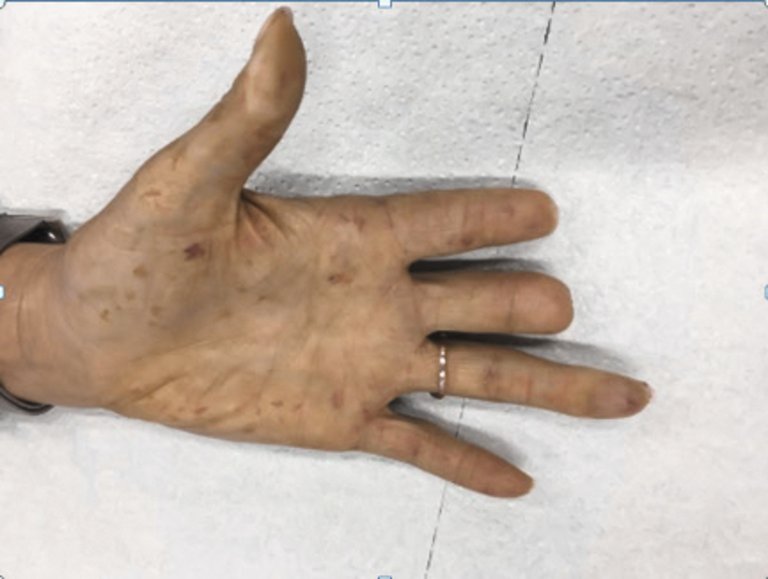
Abbildung 4: Als extreme Form der Ischämie traten bei dieser Patientin Nekrosen der Endphalangen am Zeige- und Mittelfinger auf, welche trotz vasodilatativer Therapie zu Amputationen zwangen. Die Patientin präsentiert ebenfalls typische Teleangiektasien am ganzen Integument und an den Schleimhäuten, hier palmar gut sichtbar (die Publikation erfolgt mit dem Einverständnis der Patientin).
Arthritiden oder Arthralgien sind relativ häufig und betreffen hauptsächlich die kleinen Gelenke. Der Verlauf ist, anders als bei der rheumatoiden Arthritis, meistens milde und anerosiv.
Die Mehrheit der Patienten beider Formen präsentieren Dysphagie und/oder Refluxbeschwerden relativ früh nach Krankheitsbeginn als Ausdruck der Ösophagusdysmotilität. Sie können eine ausgedehnte Motilitätsstörung des ganzen Gastrointestinaltraktes entwickeln, die bis zur Darmparese mit Malabsorption und Mangelernährung, gepaart mit einer reduzierten Lebensqualität, führen kann. Als weitere späte Komplikationen sind Ösophagusstrikturen zu erwähnen.
Das Auftreten einer Arrhythmie, einer diastolischen Dysfunktion oder Herzinsuffizienz kann auf eine kardiale Beteiligung hinweisen. Auch Perikardergüsse, meistens hämodynamisch neutral, sind häufig vorhanden. Grosse Perikardergüsse sind jedoch mit einer pulmonal-arteriellen Hypertonie und einer schlechten Prognose assoziiert.
Eine gefürchtete Komplikation bei der diffusen Systemsklerose ist die renale Krise. Vor der Ära der ACE-Hemmer war der Nierenbefall der Hauptgrund der Mortalität. Selbst unter zeitgerechtem Einsatz einer adäquaten Therapie bleibt die Überlebensprognose heutzutage bescheiden. Warnhinweise für eine mögliche Nierenbeteiligung sind ein Blutdruckanstieg (>30 mm Hg systolisch oder >20 mm Hg diastolisch), eine Verschlechterung der Nierenfunktion oder ein aktives Urinsediment. Es ist eindeutig belegt, dass eine Glukokortikoidtherapie die renale Krise begünstigen kann (vor allem bei Dosen von ≥15 mg Prednisolon-Äquivalent), sodass darauf möglichst verzichtet werden sollte.
Diagnose
Positive ANA werden in über 95% der Fälle im Serum detektiert. Von den verschiedenen Antikörpern, die üblicherweise im Rahmen der initialen Evaluation bestimmt werden, zeigen viele eine Assoziation zu gewissen Krankheitsmerkmalen. Anti-Zentromer-Antikörper sind zum Beispiel mit der limitierten Hautbeteiligung und der pulmonal-arterielle Hypertonie vergesellschaftet; die betroffenen Patienten haben hingegen ein niedrigeres Risiko, eine interstitielle Lungenerkrankung zu entwickeln. Demgegenüber sind die Scl-70-Antikörper mit der diffusen Hautbeteiligung und mit der Lungenfibrose assoziiert. Die RNA-Polymerase-III-Antikörper prädisponieren zur renalen Krise.
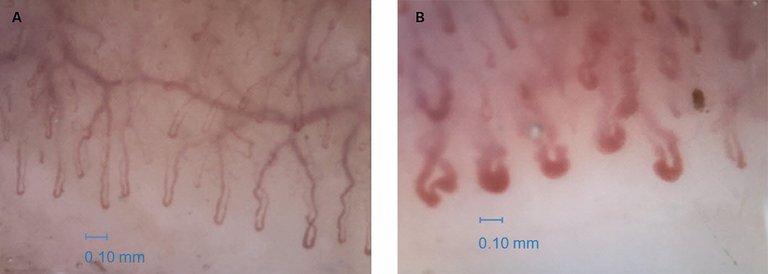
Die Kapillarmikroskopie ist eine sichere und nichtinvasive Methode zur Erfassung der Mikrovaskulopathie am Nagelfalz. Sie erlaubt in den meisten Fällen die Unterscheidung zwischen einem primären und sekundären Raynaud-Phänomen. Bei der Systemsklerose werden drei typischen Stadien der mikrovaskulären Veränderungen beschrieben:
– «early» mit einigen Megakapillaren und wenigen Blutungen (Abb. 5);
Abbildung 5: Videokapillarmikroskopie A) bei einem Gesunden mit palisadenartigem Aspekt der Kapillaren, welche meistens haarnadelförmig erscheinen; B) bei einer Patientin mit limitierter Systemsklerose, mit vielen Megakapillaren.
– «active» mit vielen Megakapillaren, vielen Blutungen und beginnendem Kapillarverlust;
– «late» mit ausgedehntem Kapillarverlust.
Interessanterweise korrelieren die Zentromer-Antikörper mit dem «active»-Muster und die Scl-70-Antikörper mit dem «late»-Muster [2]. Eine reduzierte Kapillardichte ist ein positiv prädiktiver Faktor für das Rezidiv von Fingerulzera. In diesem Zusammenhang ist jedoch zu erwähnen, dass der grösste Risikofaktor für ein solches Rezidiv stattgehabte oder aktive Ulzera darstellen. Pathologische kapillarmikroskopische Veränderungen können auch bei anderen Konnektivitiden vorkommen (klassischerweise bei der Dermatomyositis, Mischkollagenose und seltener beim systemischen Lupus erythematodes), sodass die Diagnose der Systemsklerose immer im Zusammenschau aller klinischen Elemente erfolgen sollte.
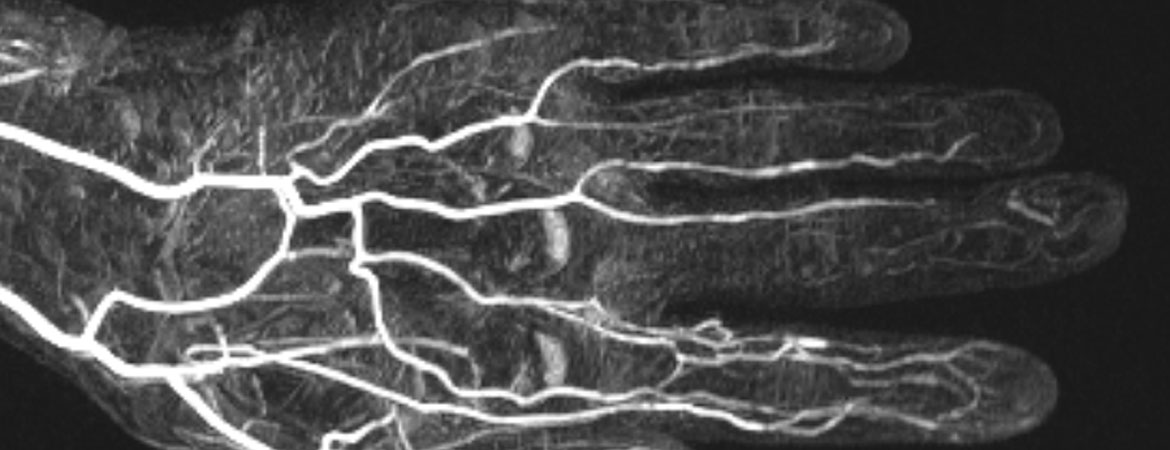
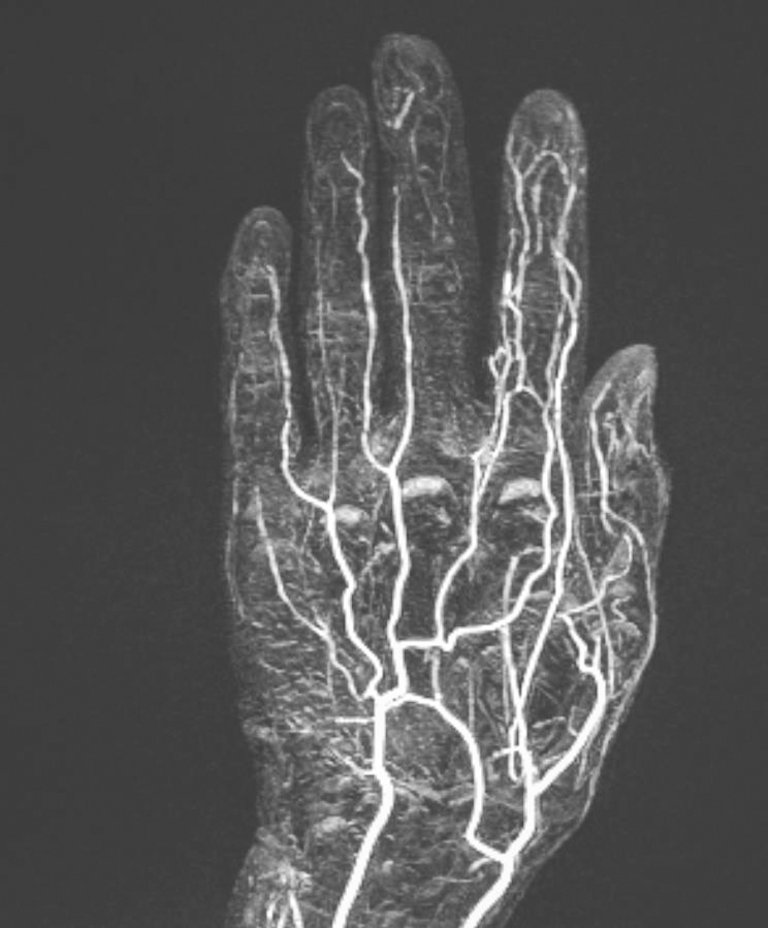
Bei unklarer akraler Ischämie kann auch die Durchführung einer MR-Angiographie zur Klärung der Ätiologie weiterhelfen. Diese spiegelt die Vaskulopathie der Digitalarterien wieder, häufig mit distalem Verschluss einhergehend (Abb. 6).
Abbildung 6: Die MR-Angiographie der linken Hand zeigt eine verminderte Kontrastierung der Digitalarterien der Strahlen III–V bei einem Patienten mit Ulkus am Mittelfinger.
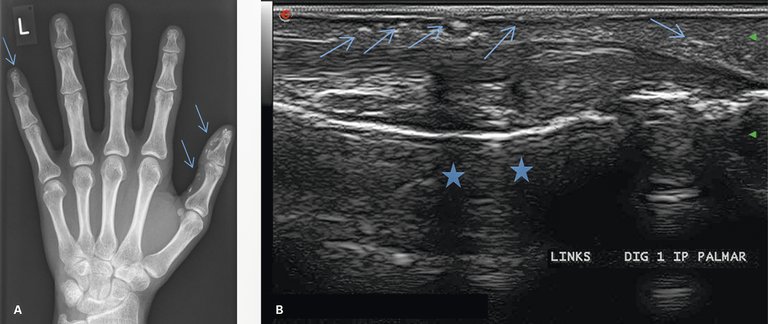
Auch anhand einer «banalen» Röntgenaufnahme der Hände kann manchmal die Diagnose einer Systemsklerose bestätigt werden, falls es eine Kalzinose oder eine akrale Osteolyse zum Vorschein bringt, wie in Abbildung 7 exemplarisch dargestellt.
Abbildung 7: Calcinosis cutis. A) Röntgen der linken Hand, Calcinosis am besten am Strahl I und V sichtbar; B) Sonographie des Daumens, palmar, bei der gleichen Patientin: die Calcinosis ist als wolkige, schalldichte Struktur (Pfeil), zum Teil mit Schallschatten (Stern) erkennbar.
Initial benötigen alle Patienten eine vollständige Lungenfunktionsprüfung (inklusive Messung der CO-Diffusionskapazität). Je nach Verlauf sollten Kontrollen alle 3–12 Monate erfolgen. Die hochauflösende Computertomographie (HRCT) der Lunge hat eine viel bessere Sensitivität für die interstitiellen Lungenveränderungen als das konventionelle Röntgen und ist demnach die Untersuchung der Wahl. Nebenbefundlich zeigt ein HRCT häufig eine Ösophagusdilatation, als unverkennbaren Beweis für dessen Dysmotilität. Klassischerweise wird jedoch mittels Ösophagus-Cinematographie (Breischluck) oder Impedanz-Manometrie danach gesucht. Letztere ist zwar invasiver, hat aber dennoch eine wesentlich bessere Sensitivität und Spezifität.
Die Echokardiographie kann indirekte Hinweise auf das Vorliegen einer pulmonal-arteriellen Hypertonie liefern, zur Bestätigung muss jedoch eine Rechtsherz-Katheteruntersuchung erfolgen.
Therapiestrategien
Da es keine krankheitsmodifizierenden Medikamente für die Systemsklerose gibt, wird eine organspezifische Behandlung erst durchgeführt, wenn ein bestimmtes Problem auftaucht (Tab. 2). Die Schwierigkeit besteht darin, das Problem rechtzeitig zu erfassen, was eine enge Zusammenarbeit zwischen den behandelnden Ärzten und eine ausführliche Patienteninformation und -schulung voraussetzt.
Tabelle 2: Therapieansätze (modifiziert nach [3]).
Kranheitsmanifestation
Therapie
Hautbefall
Hautpflege, Physiotherapie (zur Lymphdrainage, Verbesserung der Hautelastizität, Prävention/Behandlung von sekundären Gelenkkontrakturen)
ACE-Hemmer, ggf. Angiotensinrezeptor-Antagonisten und weitere Antihypertensiva
Ggf. Dialyse
Schwere, rasch progressive Form
Autologe Stammzelltransplantation
* bedarf einer Kostengutsprache in dieser Indikation
Insbesondere bei der diffusen Form sollten zu Beginn dreimonatliche Evaluationen durch das Fachteam erfolgen, bei stabilem Verlauf und vor allem bei der limitierten Form können jährliche Kontrollen allenfalls ausreichen.
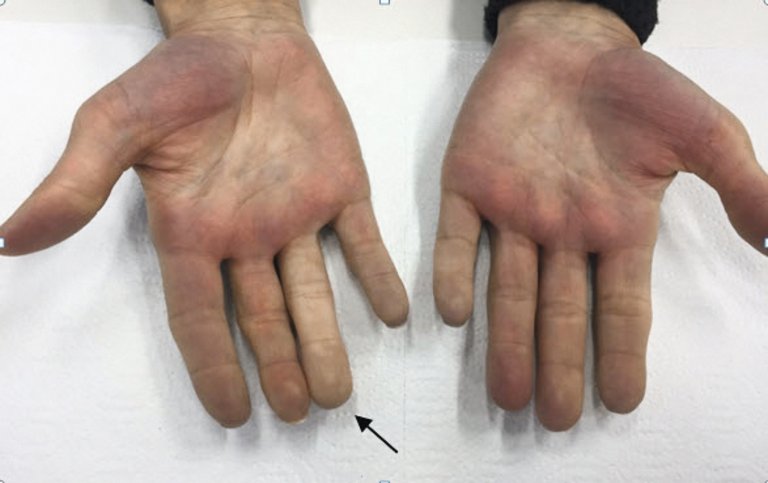
Gegen die Hautsklerose und Fingervaskulopathie werden in erster Linie nichtmedikamentöse Massnahmen wie Paraffin-Bäder, Nikotinstopp und Vermeidung von Traumatas empfohlen. Ganz zentral ist beim Raynaud-Phänomen der Kälteschutz. Der Patient sollte aber nicht nur die Extremitäten, sondern den ganzen Körper warm halten. In unserer Klinik werden die Betroffenen im Rahmen einer speziellen Sprechstunde in der Ergotherapie zu Kälteschutzmassnahmen beraten. Des Weiteren können Kalziumkanalblocker oder lokal applizierte Nitratpräparate verwendet werden. Phosphodiesterase-5-Hemmer stellen auch eine Option dar, sind aber in der Schweiz in dieser Indikation nicht kassenpflichtig. Die Kombination mit Nitratpräparaten (sei es per os oder lokal) ist aufgrund der Interaktionen verboten. Der Einsatz von Fluoxetin kann gemäss den neuen Richtlinien ebenfalls versucht werden. Gegen Fingerulzera oder kritische akrale Ischämie ist die intravenöse Gabe von Iloprost (Prostacyclinanalogon) sehr wirksam (Abb. 8). Bei ungenügender Wirkung kann Sildenafil (Phosphodiesterase-5-Hemmer) dazu verschrieben werden, bedarf jedoch einer Kostengutsprache. Zur Rezidivprophylaxe ist in der Schweiz nur Bosentan (Endothelinrezeptor-Antagonist) kassenpflichtig. Bosentan findet Verwendung insbesondere bei Patienten mit multiplen Ulzera trotz konventioneller Therapie und trägt nicht zur Heilung der Ulzera bei.
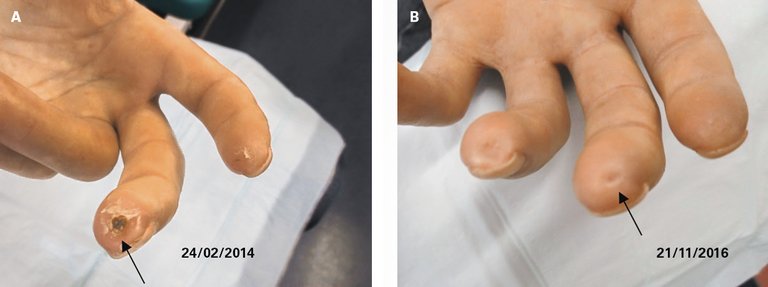
Abbildung 8: Verlauf nach vasodilatatorischer Therapie mit Illoprost.
Bei Hautbefall ist einerseits die Hautpflege von grosser Bedeutung, andererseits, bei sekundärer Problematik mit Ödem oder Tendenz zu Gelenkkontrakturen, die physio- und ergotherapeutische Behandlung. Nicht selten benötigen die Betroffenen zur Erhaltung einer möglichst guten Beweglichkeit, insbesondere der Hand- und Fingergelenke, eine regelmässige, oft wöchentliche Therapie. Zur Verbesserung der Beweglichkeit können auch selbständig durchgeführte Dehnungsübungen beitragen. Im Verlauf können Patienten eine spontane Besserung der Hautveränderungen erleben. Oft bleiben die bereits vorhandenen Kontrakturen aber bestehen. Der Einsatz von Methotrexat ist bei der diffusen Systemsklerose im Frühstadium empfohlen. Bei rapid progressivem Verlauf sollte trotz hoher Toxizität auch die Gabe von Cyclophosphamid erwogen werden.
Gegen die Sicca-Symptomatik, welche wie beim primären Sjögren-Syndrom stark beeinträchtigen kann, sind lokale Massnahmen angebracht. Pilocarpin-Tropfen oder -tabletten (Salagen®) können für die Xerophthalmie ebenfalls von Nutzen sein. Es sollte auf eine gute Zahnhygiene geachtet werden. Oft übernimmt die Krankenkasse auf Anfrage einen Teil der dentalhygienischen Behandlung.
Bei der gastrointestinalen Beteiligung können im Frühstadium Prokinetika eingesetzt werden. Im fortgeschrittenen Stadium wirken diese aufgrund der zunehmenden Fibrosierung hingegen nicht mehr. Grundsätzlich sollten alle Patienten eine Behandlung mit Protonenpumpen-Inhibitoren (PPI) erhalten, weil dadurch der gastroösophageale Reflux, die sekundär entstandenen distalen Ösophagusstrikturen und die spätere Bougierung der letzteren verhindert werden können. Bei rezidivierender Diarrhoe kann man in Annahme einer bakteriellen Überwucherung entsprechend antibiotisch behandeln. Bei Hinweisen auf therapieresistente schwere Malabsorption mit Kachexie soll eine parenterale Ernährung in Betracht gezogen werden. Diese kann stationär eingeleitet und unter regelmässigem Monitoring zuhause weitergeführt werden. Damit gewinnen die Patienten an Lebensjahren und -qualität.
Viele Fortschritte wurden in den letzten Jahren im Bereich der Therapie der Lungenpathologie gemacht, sei es für die pulmonal-arterielle Hypertonie oder die interstitielle Lungenerkrankung.
Bei der pulmonal-arteriellen Hypertonie muss hervorgehoben werden, dass eine frühzeitige Diagnosestellung selbst bei asymptomatischen Patienten prognostisch günstig ist. Dies unterstreicht die Wichtigkeit des regelmässigen Screening auf pulmonal-arterielle Hypertonie. Bis jetzt wurden therapeutisch in erster Linie Endothelinrezeptor-Antagonisten und Phosphodiesterase-Inhibitoren eingesetzt, in schwierigen Fällen ein parenteral appliziertes Prostazyklin-Analogon dazu. Seit zwei Jahren steht uns auch Riociguat (ein Guanylatzyklase-Stimulator) in dieser Indikation zur Verfügung. Neu zugelassen in der Schweiz ist Selexipag – der erste Prostazyklinrezeptor-Agonist. Selexipag hat gegenüber den Prostazyklin-Analoga den grossen Vorteil der oralen Applikationsform, die Auftitrierung bedarf allerdings eines häufigen Monitoring und einer guten Patientenaufklärung [4]. Neuere Studien zeigen, dass eine Kombinationstherapie bereits in frühen Stadien möglicherweise der Monotherapie überlegen ist. Der Therapieentscheid bei pulmonal-artierieller Hypertonie gehört idealerweise in die Hände eines interdisziplinären Teams von Rheumatologen, Pneumologen und Kardiologen.
Bei der interstitiellen Lungenerkrankung sollte in erster Linie Cyclophosphamid eingesetzt werden. Dank neuer Daten kann auch die Gabe von Mycophenolat mofetil in Erwägung gezogen werden. Der Therapieeffekt wurde in einer kürzlich erschienen Studie als mit Cyclophosphamid vergleichbar beurteilt. Das Nebenwirkungsprofil favorisierte jedoch das Mycophenolat mofetil. Pirfenidon, ein antifibrotischer Wirkstoff, der bereits bei der idiopathischen Lungenfibrose zum Einsatz kommt, wurde vor Kurzem in Bezug auf die Verträglichkeit bei Systemsklerose-Patienten untersucht. Die Daten erlaubten jedoch keinen Rückschluss auf den therapeutischen Effekt. Da der gastroösophageale Reflux die interstitielle Lungenerkrankung ungünstig beeinflusst, ist eine PPI-Behandlung bei interstitieller Lungenerkrankung obligat.
Die renale Krise ist ein Notfall, die Behandlung muss rasch unter stationären Bedingungen erfolgen. ACE-Hemmer sind die erste Therapieoption, da sich die Überlebensrate bei Nierenbefall seit Einführung dieser Behandlung massiv verbessert hat. Es empfiehlt sich deswegen, auch bei Hinweisen auf nur leicht hypertone Blutdruckwerte bevorzugt ACE-Hemmer einzusetzen. Manchmal wird der Patient trotz aller Bestrebungen jedoch dialysepflichtig.
Bei rasch progredienter Erkrankung kann in ausgewählten Fällen eine autologe Stammzelltransplantation diskutiert werden (Tab. 2).
Prognose
Die Prognose ist sehr variabel. Die limitierte Form geht grundsätzlich mit einer besseren Prognose einher. Das Vorliegen eines Organbefalls (Lunge, Niere, Herz) und das männliches Geschlecht sind mit einer schlechteren Prognose vergesellschaftet.
Ausblick
Antifibrotische Mittel werden dringend gebraucht. Nintedanib (Tyrosinkinase-Inhibitor) und Pirfenidon werden bei der idiopathischen Lungenfibrose eingesetzt und scheinen auch bei der interstitiellen Lungenerkrankung im Rahmen der Systemsklerose vielversprechend zu sein. Tocilizumab zeigte in einem Patientenkollektiv mit erhöhten Entzündungsparametern einen Trend zur Besserung der Hautveränderungen. Ebenfalls bewirkte Rituximab in kleinen Studien eine leichte Verbesserung der Hautveränderungen und eine leichte Verbesserung respektive Stabilisierung der Lungenfunktionstests. Weitere Studien zum therapeutischen Effekt dieser Substanzen bei der Systemsklerose sind geplant oder bereits im Gange, in den nächsten Monaten oder Jahren dürfen wir mit deren Ergebnissen rechnen [5].
Das Wichtigste für die Praxis
• Bei neu aufgetretenem oder sich verschlechterndem Raynaud-Phänomen oder unerklärlicher Fingerschwellung ist es sinnvoll, die antinukleären Antikörper (ANA) zu bestimmen und die Patienten niederschwellig zur weiteren Abklärung zu überweisen.
• Regelmässige, initial monatliche Kontrolle von Urinstatus und Blutdruck durch die Patienten selber und den Hausarzt sind sehr wichtig. Bei Verdacht auf eine renale Krise sollte eine unverzügliche Zuweisung erfolgen.
• Das Screening auf pulmonal-arterielle Hypertonie sollte konsequent durchgeführt werden, weil die rechtzeitige Behandlung mit einer besseren Prognose assoziiert ist. Eine Kombinationstherapie ist früh in Erwägung zu ziehen.
• Männliches Geschlecht, die diffuse Form und das Vorhandensein eines Organbefalls sind mit einer schlechten Prognose assoziiert.
• Ein regelmässiges (mindestens jährliches) Monitoring durch ein erfahrenes Fachteam ist empfohlen, auch bei asymptomatischen Patienten, um den Einsatz einer zeitgerechten Therapie zu gewährleisten
Verdankung
Für die kritische Durchsicht des Manuskripts und die konstruktiven Anregungen möchte ich mich bei Prof. Dr. med. Peter Villiger, Universitätsklinik für Rheumatologie, Immunologie und Allergologie, Inselspital, Bern, und bei Frau Dr. med. Magdalena Gantenbein, Fachärztin für Allgemeine Innere Medizin, Basel, ganz herzlich bedanken.
Disclosure statement
Die Autorin hat keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Korrespondenz
Dr. med. Diana Dan Service de Rhumatologie, Hôpital orthopédique CHUV Avenue Pierre-Decker CH-1011 Lausanne diana.dan-rusei[at]chuv.ch
Literatur
1 Dan D. Die Systemsklerose. Der informierte Arzt. 2012;2:14–5.
2 Cutolo M, Editor. Atlas of capillaroscopy in rheumatic diseases. 1st ed. Milano: Elsevier;2010.
3 Villiger P. Systemsklerose. Therapeutische Umschau. 2008;65:283–7.
4 Kowal-Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2016;0:1–13.
5 Khanna D, Distler JHW, Sandner P, Distler O. Emerging strategies for treatment of systemic sclerosis. Journal of Scleroderma and Related Disorders. 2016;1(2):186–93.