


Publiziert am 07.03.2018
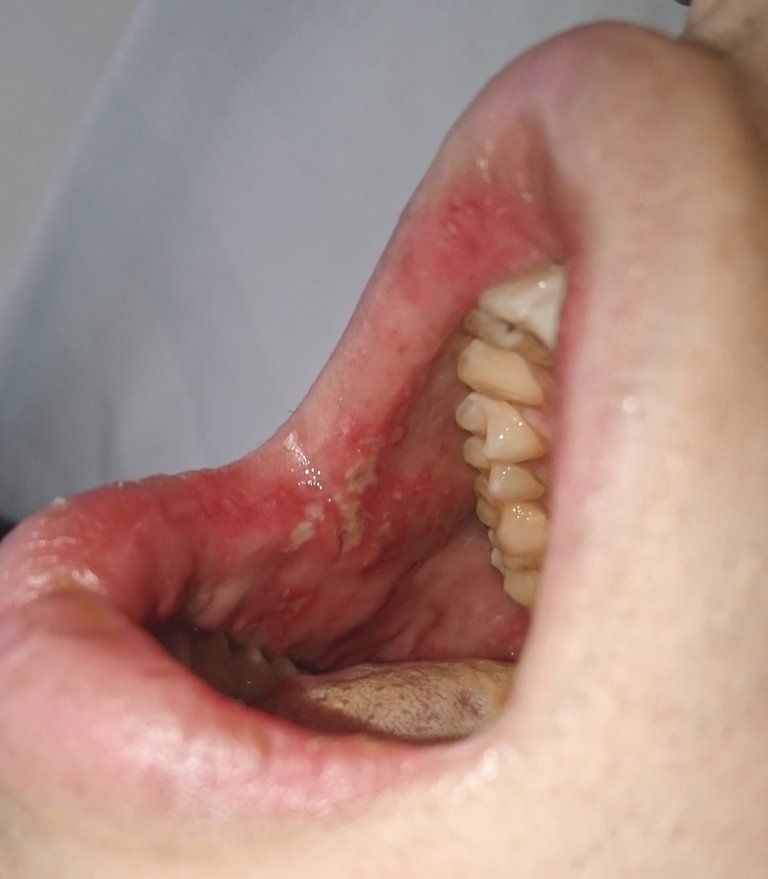
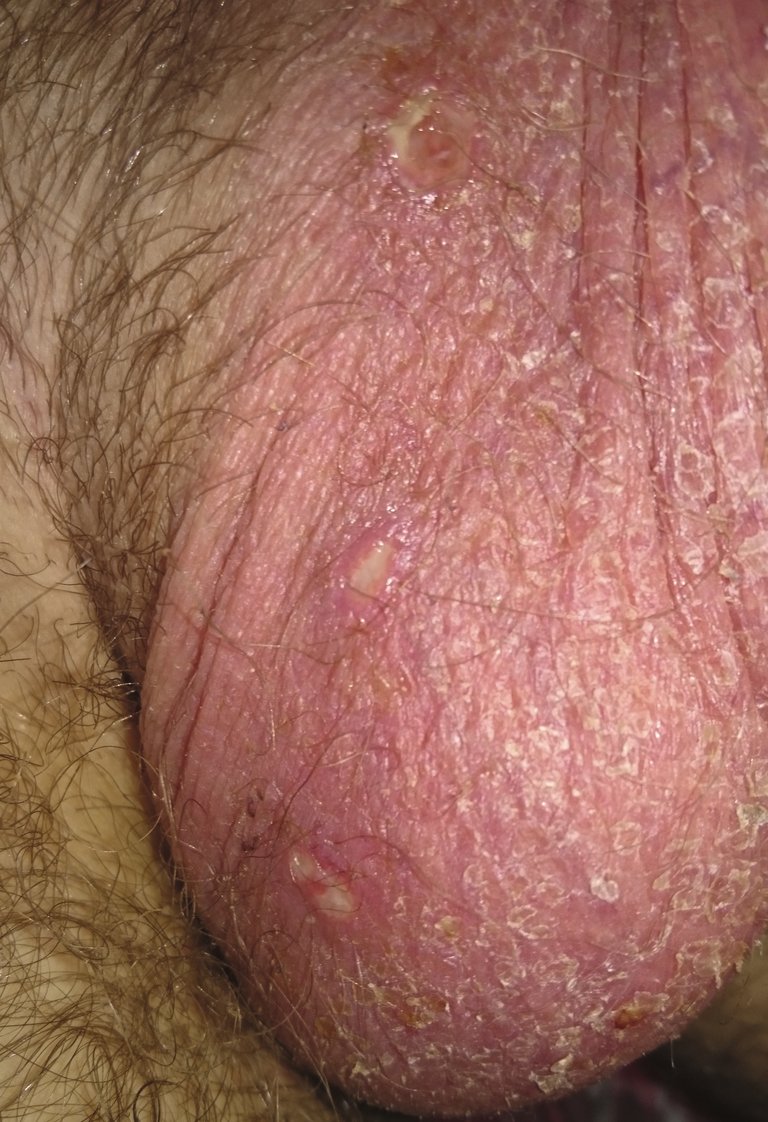
| Tabelle 1: Diagnosekriterien des Morbus Behçet. | |
| Diagnostische Kriterien nach der «International Study Group for Behcet’s Disease» | |
| Obligates Symptom: | Rezidivierende orale Aphthen (mindestens 3-mal jährlich) |
| Plus 2 der 4 folgenden Kriterien: | Rezidivierende Genitalulzera |
| Typische Augenläsionen (Uveitis anterior, Uveitis posterior, retinale Vaskulitis) | |
| Hautveränderungen (Erythema nodosum, Follikulitis, sterile Pusteln) | |
| Positiver Pathergie-Test | |
Veröffentlicht unter der Copyright-Lizenz.
"Attribution - Non-Commercial - NoDerivatives 4.0"
Keine kommerzielle Weiterverwendung ohne Genehmigung.
See: emh.ch/en/emh/rights-and-licences/
