Inselspital Bern; a Universitätsklinik für Allgemeine Innere Medizin, b Universitätsklinik für Infektiologie und Reisemedizin, c Universitätsinstitut für Diagnostische, Interventionelle und Pädiatrische Radiologie
Eine 37-jährige Patientin wurde notfallmässig vom Hausarzt mit Epistaxis, multiplen Hämatomen und progredienter Thrombozytopenie zugewiesen.
Hintergrund
Die Immunthrombozytopenie (ITP) gehört zu den häufigsten Ursachen einer Thrombozytopenie bei ansonsten asymptomatischen Erwachsenen. Die jährliche Inzidenz in Europa beträgt bei Erwachsenen ein bis fünf Personen pro 100 000 [1–3]. Studien aus den USA zeigen eine etwa doppelt so hohe Prävalenz bei Erwachsenen (zwölf pro 10 000), was auf den chronischen Verlauf zurückgeführt werden kann [4]. Rund 20–30% der neu diagnostizierten Patienten sind zum Zeitpunkt der Diagnose asymptomatisch; es handelt sich meist um einen zufälligen Laborbefund [5, 6]. Klinisch wird zwischen einer primären und einer sekundären ITP unterschieden. In den meisten Fällen (60–70%) liegt eine primäre ITP vor. Sie basiert auf spezifischen IgG-Antikörpern, die an Glykoproteine der Thrombozyten (Tc) binden. In den restlichen Fällen handelt es sich um eine sekundäre ITP im Rahmen von Autoimmunerkrankungen (etwa systemischer Lupus erythematodes), hämatologischen Erkrankungen (chronische lymphatische Leukämie, Hodgkin Lymphom), Immunmangelsyndromen, Infektionen (wie HIV, Hepatitis C, Helicobacter pylori) oder medikamentös-toxischen Ursachen [7, 8]. Die ITP kann in eine akute Form (Dauer bis drei Monate), eine persistierende Form (Dauer drei bis zwölf Monate) und eine chronische Form (Dauer mehr als zwölf Monate) klassifiziert werden. Die persistierende ITP ist häufiger bei Kindern und die chronische bei Erwachsenen anzutreffen [9].
Fallbericht
Anamnese
Die 37-jährige Patientin wurde notfallmässig vom Hausarzt mit Epistaxis, multiplen Hämatomen und progredienter Thrombozytopenie zugewiesen. Des Weiteren klagte die Patientin über Inappetenz und Nachtschweiss, sowie seit dem Vortag über unproduktiven Husten. Wegen seit vier Wochen bestehenden abdominalen und thorakolumbalen Schmerzen war ambulant bereits eine Abdomensonographie durchgeführt worden. Hier zeigten sich eine Splenomegalie sowie zwei vergrösserte Lymphknoten im Bereich des Leberhilus. Die Patientin stammt aus Eritrea und ist vor vier Jahren in die Schweiz gekommen. Zum damaligen Zeitpunkt wurde bei Husten mit Hämoptyse eine Sputum-Kultur angesetzt, in der keine Mykobakterien nachgewiesen werden konnten.
Status und Befunde
Die Patientin präsentierte sich in reduziertem Allgemeinzustand, afebril und hämodynamisch stabil. An den unteren Extremitäten und am rechten Oberarm zeigten sich multiple Hämatome, sowie mehrere tastbare, indolente Lymphknoten zervikal und inguinal. Die Schleimhäute waren unauffällig.
Laboranalytisch zeigte sich ein pathologisches Blutbild ohne Thrombozyten, mit einer Lymphopenie, einer normochromen, normozytären Anämie sowie eine Mikrohämaturie. Der Gerinnungsstatus sowie die Leberwerte fielen unauffällig aus.
Diagnose
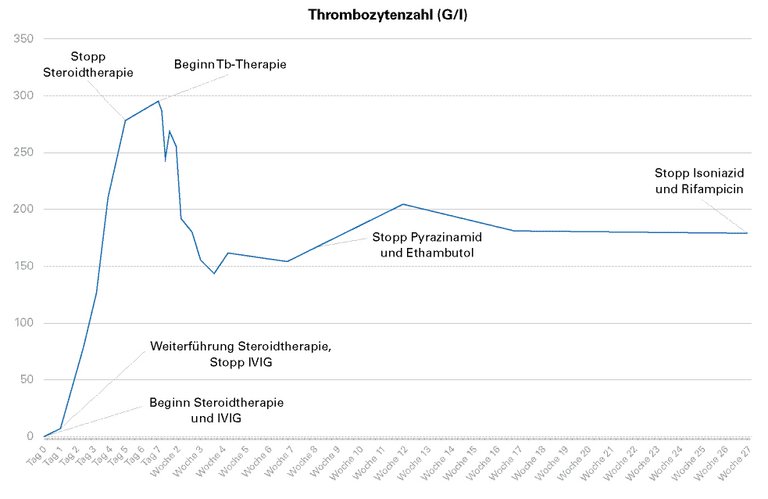
Als nach Gabe eines Thrombozytenkonzentrates kein Anstieg der Tc erfolgte, wurde die Verdachtsdiagnose einer ITP gestellt. Aufgrund der akuten Blutung wurde sofort eine Therapie mit Prednisolon und intravenösen Immunglobulinen (IVIG) eingeleitet. Es kam zu einem deutlichen Anstieg und schliesslich zu einer Normalisierung der Tc (Abb. 1), so dass die IVIG nach zwei Gaben gestoppt werden konnte.
Abbildung 1: Verlauf der Thrombozytenzahl. IVIG = intravenöse Immunglobuline.
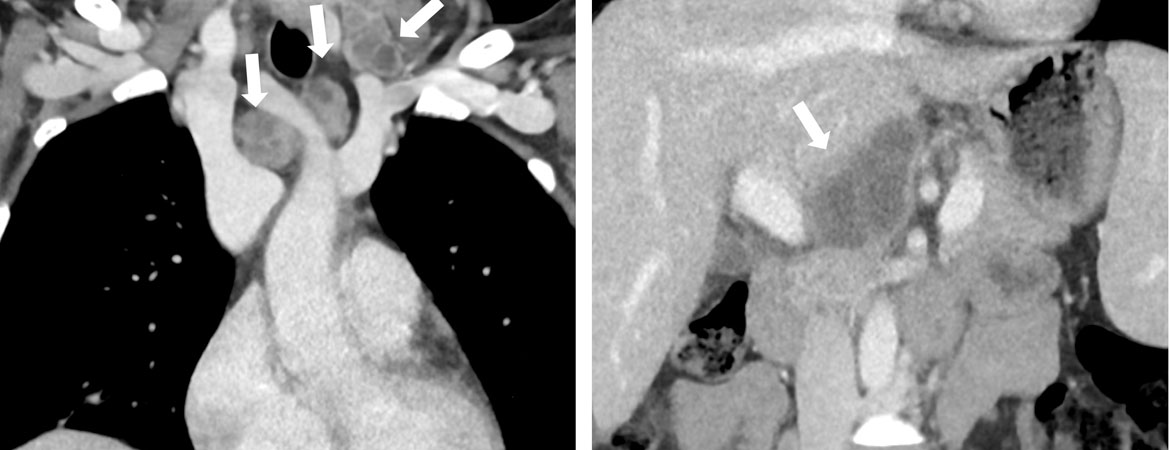
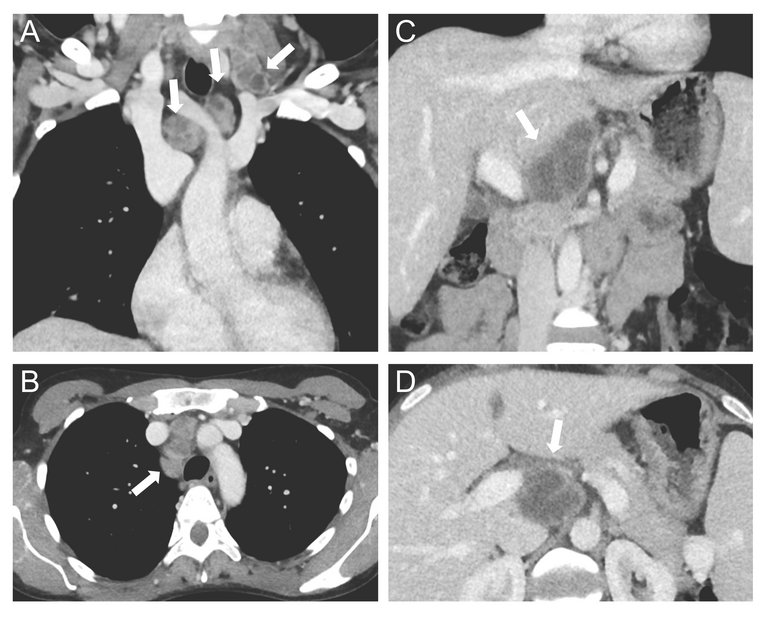
Es gab keine Hinweise auf eine Autoimmunerkrankung, ein Antiphospholipid-Syndrom, eine Infektion mit dem humanen Immundefizienz-Virus (HIV), eine virale Hepatitis oder eine Paraproteinämie als mögliche sekundäre Ursachen. Ein positives Helicobacter- pylori (H. pylori)-Antigen im Stuhl war hinweisend auf eine Helicobacter-pylori-Infektion. Die Computertomographie (CT) des Thorax und Abdomens zeigte eine zervikale, supraklavikuläre, mediastinale und hiläre Lymphadenopathie mit zentral nekrotischen Lymphknoten und ein Infiltrat im Mittellappen, sowie eine leichte Splenomegalie (13,5 × 12 × 4,5 cm) ohne fokale Läsionen und ein normales Leberparenchym (Abb. 2). Aufgrund der Symptome und des auffälligen CT wurden erneut eine Sputumuntersuchung durchgeführt. Hier zeigten sich mikroskopisch säurefeste Stäbchen. Die «polymerase chain reaction» (PCR) war positiv für Mycobacterium tuberculosis. Damit konnte die Diagnose einer offenen Tuberkulose gestellt werden.
Abbildung 2: Computertomographie mit koronarer (A , C) und axialer Reformation (B , D) des Thorax und Oberbauchs. Zervikale, mediastinale und retroperitoneale Lymphknoten mit zentraler Nekrose sind mit Pfeilen gekennzeichnet.
Therapie und Verlauf
Nach Normalisierung der Tc-Zahl am vierten Tag und bei stabilem Verlauf wurde die Steroidtherapie am fünften Tag gestoppt. In der GenXpert-Untersuchung wurde eine Rifampicin-Resistenz ausgeschlossen und eine tuberkulostatische Therapie mit Isoniazid, Rifampicin, Pyrazinamid und Ethambutol begonnen. Die weitere Diagnostik zeigte ebenfalls keine Resistenz gegenüber den anderen Antibiotika. Der Guideline entsprechend wurde nach zwei Monaten Pyrazinamid und Ethambutol gestoppt. Die Therapie mit Isoniazid und Rifampicin wurde für weitere vier Monate fortgesetzt. Die Tc blieben stets im Normbereich (Abb. 1). Die initial zusätzlich bestehende Lymphopenie und Anämie normalisierten sich unter der tuberkulostatischen Therapie vollständig. Im Verlauf wurde eine Eradikationstherapie für den Helicobacter pylori durchgeführt.
Diskussion
In Zusammenschau der Befunde halten wir bei dieser Patientin eine sekundäre ITP im Rahmen einer offenen Tuberkulose für die wahrscheinlichste Krankheitserklärung. Unterstützt wird diese Diagnose durch die Tatsache, dass die Tc-Zahl nach Einleitung der tuberkulostatischen Therapie (Tag 7) und trotz Sistieren der Steroidtherapie (Tag 5) und der Therapie mit IVIG (Tag 1) im Normbereich geblieben ist. Bei Patienten mit einer primären ITP wäre ein langfristiges Ansprechen nach einer Langzeitsteroidtherpie über mehrere Wochen zu erwarten. Bei der sekundären ITP ist die H.-pylori-Infektion als Differentialdiagnose nicht ausgeschlossen, aber wenig wahrscheinlich, weil die Eradikationstherapie erst einen Monat später im ambulanten Setting begonnen wurde und sich bis dahin kein Rezidiv der Thrombozytopenie trotz Sistieren der Steroid- und IVIG-Therapie gezeigt hatte.
Als Ursache der Splenomegalie käme bei fehlenden laborchemischen und radiologischen Hinweisen einer Hepatopathie auch die Tuberkulose in Frage. Bei jedoch nur leichter Splenomegalie ohne fokale Läsionen gingen wir von der Immunthrombozytopenie als Ätiologie der Splenomegalie aus.
Neben genetischer Prädisposition werden bei der primären ITP spezifische IgG-Antikörper durch die B-Zellen des Patienten produziert. Diese binden an bestimmte Glykoproteine der Thrombozyten (GPIIb/IIIa und GP1b/IX/V), was zur Folge hat, dass die Tc entweder durch Makrophagen innerhalb des retikuloendothelialen Systems [8, 11] oder durch eine Dysregulation der T-Zellen [8] zerstört werden. Bei einer sekundären ITP, wie sie bei unserer Patientin vorlag, werden in der Regel Antikörper gegen Tc-Glykoproteine durch ein molekulares Mimikry der infektiösen Komponenten (etwa virale Antigene) oder Medikamente produziert [11]. Weitere sekundäre ITP bei autoimmunen oder hämatologischen Erkrankungen sind aufgrund von Veränderungen der Immunhomöostase mit Bildung von Autoantikörpern beschrieben [12–14].
Die klinische Präsentation kann von asymptomatischen Verläufen über Petechien, Purpura, Nasenbluten, Hämaturie bis zu lebensbedrohlichen Hämorrhagien variieren [9, 15]. Die ITP ist eine Ausschlussdiagnose bei Patienten mit isolierter Thrombozytopenie [8]. Neben einer ausführlichen Anamnese und klinischen Untersuchung gehören die Untersuchung des Blutabstriches und die Suche nach sekundären Ursachen zur Abklärung (etwa für HIV, Hepatitis-C-Virus, H. pylori, Immunserologie, Immunglobulin im Serum) [16]. Die Bestimmung der Tc-Antikörper ist wegen niedriger Sensitivität und Spezifität nicht empfohlen.
Eine Tuberkulose ist eine seltene Ursache der ITP. Bis 2016 wurden bislang lediglich 50 Fälle publiziert [17]. Der erste Fall wurde in den 1950er Jahren beschrieben [18]. In einer retrospektiven Studie von 1223 Patienten in Taiwan mit der Diagnose einer Tuberkulose zwischen 2000 und 2013 fand sich in 33 Fällen (2,6%) eine ITP [18]. In der Analyse der 50 publizierten Fällen wurde ein kausaler oder wahrscheinlicher Zusammenhang bei 34 (68%) postuliert, da sich die ITP erst nach Beginn der tuberkulostatischen Therapie besserte oder die tuberkulostatische Therapie mit der ITP Therapie begonnen wurde [17]. Der pathophysiologische Mechanismus der TB-assoziierten ITP ist unklar. Die Bildung von Anti-Tc-Antikörpern im Rahmen der Immunabwehr gegen Mycobacterium tuberculosis wird diskutiert, wobei Anti-Tc-Antikörper nur in wenigen Fällen detektiert wurden [17]. Bei Patienten mit Immunerkrankungen (Morbus Addison, Morbus Crohn, Colitis ulcerosa, Sprue, Spondylitis ankylosans, systemischer Lupus erythematodes, Sjögren Syndrom, Sklerodermie, Derma- und Polyomyositis, Psoriasis, Polyarteritis nodosa, autoimmun hämolytische Anämie, Thyreoiditis Hashimoto, Myasthenia gravis und ITP) besteht aufgrund der Immundysfunktion oder immunsuppressiven Therapie ein erhöhtes Risiko für TB [19]. Die in der Studie beobachtete höhere Prävalenz einer TB bei Patienten mit Immunerkrankungen kann aber auch Folge der intensiveren Testung für eine TB vor Beginn einer immunsupprimierenden Therapie sein.
Die Therapie einer ITP bei akuter Blutung beinhaltetet eine intravenöse Steroidtherapie in Kombination mit IVIG, Tranexamsäure und Tc-Transfusionen [20]. Thrombopoietin-Rezeptor-Agonisten (TPO-RAs) wurden ebenfalls angewendet [20]. Bei fehlender Blutung ist die Therapie der ersten Wahl orale Glukokortikoide (Prednison 1–2 mg/kg pro Tag über mehrere Wochen) oder Dexamethason (40 mg pro Tag für vier Tage monatlich je nach Tc-Zahl) plus die intravenöse Gabe von Immunglobulinen (IVIG)- oder Anti-D Immunglobulinen (die letzte für Rhesus (D) positive Patienten). Unter Steroidtherapie ist ein positives Ansprechen in 70–80% zu erwarten, allerdings bei geringer langfristiger Remissionsrate [21]. Bei persistierender, schwerer Thrombozytopenie war eine Splenektomie über Jahrzehnte die Therapie der Wahl als Zweitlinientherapie mit einer Remissionsrate von 60–70% nach fünf Jahren [8]. Trotz diesem Erfolg unterziehen sich weniger als 25% der Patienten einer Splenektomie aufgrund der Komplikationen (postoperative Komplikationen, erhöhtes Infekt- und Thromboserisiko, pulmonale Hypertonie) [8]. Zudem steht mit Rituximab heute eine ebenfalls effektive Zweitlinientherapie zur Verfügung. Bei Patienten, die auf die Erstlinientherapie nicht ansprachen, zeigte sich ein initiales Ansprechen auf die Rituximab-Monotherapie in 40–60% der Fälle mit einer Rezidivrate von 80% nach fünf Jahren [8]. In Kombination mit Glukokortikoiden führte die Therapie mit Rituximab zu einer anhaltenden Remission von 63% nach sechs Monaten [22]. Die Gabe der TPO-RAs-Romiplostim und Eltrombopag führte sowohl zu kurz- als auch langfristigen Therapieerfolgen von >80% [8].
Bei Fällen mit einer ITP und einer aktiven Tuberkulose wurde meist eine Therapie mit Glukokortikoiden und IVIG durchgeführt [17]. Dabei zeigte sich häufig ein sehr schlechtes Ansprechen und in einigen Fällen war die ITP erst nach Beginn der tuberkulostatischen Therapie erfolgreich behandelbar. Ein relevanter Tc-Anstieg erfolgte bei 60% der Patienten innerhalb von einem Monat, in einem Fall erst nach drei Monaten. In einigen Fällen mit asymptomatischer ITP stiegen die Tc nach Beginn der tuberkulostatischen Therapie an und normalisierten sich. Bei einer symptomatischen ITP ist die Kombination der TB-Therapie mit IVIG und Glukokortikoiden nötig [17].
Das Wichtigste für die Praxis
• Die Immunthrombozytopenie (ITP) ist eine wichtige Ursache einer Thrombozytopenie. Ihr liegen unterschiedliche Ätiologien zugrunde.
• Im Rahmen der Abklärung einer ITP sollte auch an seltene, infektiöse Ursachen gedacht werden.
• Die Tuberkulose ist mit 500 bis 600 Fällen pro Jahr in der Schweiz nach wie vor eine eher seltene Erkrankung. Aufgrund der zunehmenden Reisetätigkeit und der Migration ist die Tuberkulose eine wichtige Differentialdiagnose.
Disclosure statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Korrespondenz
Thrasyvoulos Gkrezios, dipl. Arzt Universitätsklinik für Viszerale Chirurgie und Medizin Inselspital Freiburgstrasse 18 CH-3010 Bern thrasyvoulos.gkrezios[at]insel.ch
Literatur
1 Frederiksen H, Schmidt K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood. 1999;94(3):909.
2 Terrell DR, Beebe LA, Vesely SK, Neas BR, Segal JB, George JN. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol. 2010;85(3):174.
3 Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol. 2009;83(2):83–9. Epub 2009 Feb 23.
4 Terrell DR, Beebe LA, Neas BR, Vesely SK, Segal JB, George JN. Prevalence of primary immune thrombocytopenia in Oklahoma. Am J Hematol. 2012. Sep;87(9):848–52. Epub 2012 Jun 5.
5 Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood. 2001;97(9):2549.
6 Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR, Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol. 2003;122(6):966.
7 Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386.
8 Lambert M, Gernscheimer T. Clinical updates in adult immune thrombocytopenia. Blood. 2017;129:2829–35.
9 Swinkels M, Rijkers M, Voorberg J, Vidarsson G, Leebeek FWG, Jansen AJG. Emerging Concepts in Immune Thrombocytopenia. Front. Immunol. 2018;9:880.
10 Stasi R, Provan D. Helicobacter pylori and Chronic ITP. Hematology Am Soc Hematol Educ Program. 2008.
11 Audia S, Mahévas M, Samson M, Codeau B, Bonnotte B. Pathogenesis of immune thrombocytopenia. Autoimmunity Reviews. 2017;16:620–32.
12 Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood. 2009;113(26):6511. Epub 2009 Apr 24.
13 Mahévas M, Chiche L, Uzunhan Y, Khellaf M, Morin AS, Le Guenno G, Péronne V, et al. Association of sarcoidosis and immune thrombocytopenia: presentation and outcome in a series of 20 patients. Medicine (Baltimore). 2011;90(4):269.
14 Cines DB, Liebman H, Stasi R. Pathobiology of secondary immune thrombocytopenia. Semin Hematol. 2009;46(1 Suppl 2):S2.
15 Neunert C, Noroozi N, Norman G, Buchanan GR, Goy J, Nazi I, et al. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J Thromb Haemost. 2015;13(3):457. Epub 2015 Jan 14.
16 Neunert C, Lim W, Crowther M, Cohen A, Solberg L Jr, Crowther MA. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117(16):4190.
17 Weber S, Bélard S, Rai S, Reddy S, Belurkar S, Saravu S. Immune thrombocytopenia secondary to tuberculosis: a case and review of literature. Int J Tuberc Lung D. 2017;21(4):466–70.
18 Shu-Ruei Wu, Hsiao-Ching Kuo. Awareness of tuberculosis among patients with newly diagnosed immune thrombocytopenia in the endemic area. Ann Hematol. 2017;96:1773–4.
19 Ramagopalan S, Goldacre R, Skingsley A, Conlon C, Goldcare M. Associations between selected immune-mediated diseases und tuberculosis: record-linkage stuidies. BMC Medicine. 2013;11:97.
20 Cooper N. State of the art – how I manage immune thrombocytopenia. British Journal of Hematology. 2017;177:39–54.
21 Neunert C. Management of newly diagnosed immune thrombocytopenia: can we change outcomes? Blood advances. 2017;1(24):2295–301.
22 Zaja F, Bacarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood. 2010;115(14):2755–62.