a Klinik für Innere Medizin und Nephrologie, Hirslanden Klinik St. Anna, Luzern; b Kempf und Pfaltz, Histologische Diagnostik AG, Zürich; c Onkozentrum Luzern, Hirslanden Klinik St. Anna, Luzern; d Klinik für Transplantationsimmunologie und Nephrologie, Universitätsspital Basel
Beim klinischen Bild von Ulcera cruris kommen viele Differentialdiagnosen infrage (siehe Tab. 1). Zu den seltenen Ursachen zählen Lymphome mit kutaner Manifestation. In diesem Fallbeispiel geht es um die Diagnose eines Epstein-Barr-Virus-(EBV-)positiven, diffusen, grosszelligen B-Zell-Lymphoms («diffuse large B-cell lymphoma» [DLBCL]) mit primär kutaner Manifestation und Ulzerationen.
Tabelle 1: Differentialdiagnosen eines Ulcus cruris (modifizierte Tabelle) [1–3, 5].
Vaskuläre Ursachen
Periphere arterielle Verschlusskrankheit
Chronisch venöse Insuffizienz
Vaskulitis
Mikroangiopathie mit/ohne Neuropathie bei Diabetes mellitus
Die hausärztliche Zuweisung der 76-jährigen Patientin erfolgte zur Abklärung von seit etwa acht Wochen bestehenden Ulcera cruris beidseits. Anamnestisch war bis auf einen ausgeprägten Nachtschweiss keine B-Symptomatik eruierbar. Aktenanamnestisch präsentierten sich zwei Monate zuvor die unteren Extremitäten beidseits umfanggleich mit distal betonter, schmerzloser Fuss- und Unterschenkelschwellung und eindrückbaren Ödemen; inguinal konnten des Weiteren indolente, gut verschiebliche Lymphknoten getastet werden. Unter einer Kompressionstherapie kam es an beiden Unterschenkeln zu einer flächenhaften, teils indurierten Rötung/Hyperpigmentierung der Haut. Im Verlauf entwickelten sich aus den schmerzlosen Hautindurationen offene Läsionen: unter anderem eine grössere, prätibiale Läsion links und eine Läsion medial an der rechten Wade mit Entwicklung eines floriden, grösseren Ulkus mit Fibrinbelag und nekrotischen Wundrändern.
In der Duplexsonographie fanden sich keine Hinweise für eine Phlebothrombose. Eine relevante Makroangiopathie konnte ebenfalls nicht festgestellt werden (Knöchel-Arm-Index: rechts 1,21, links 1,33).
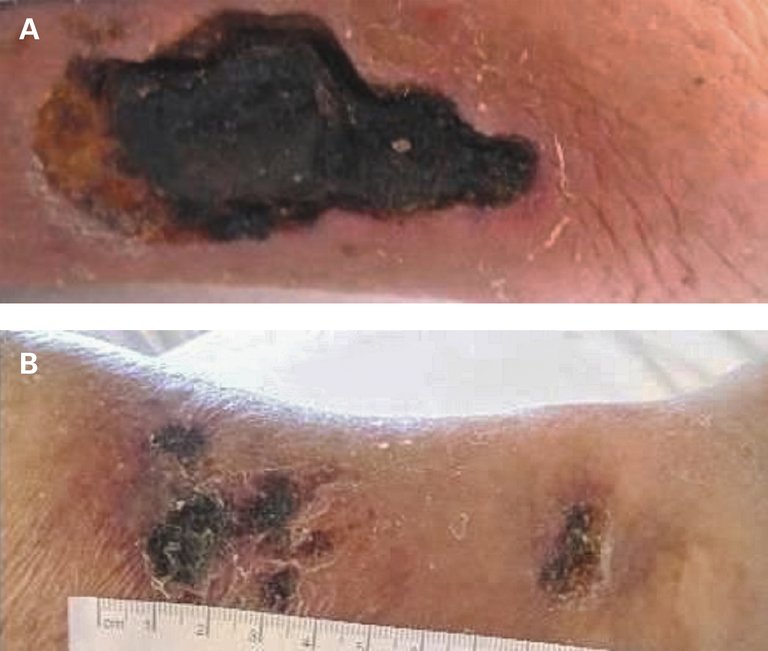
Bei laborchemisch erhöhtem C-reaktivem Protein (CRP: 21,9 mg/l, Norm: <5,0 mg/l) und erhöhten antinukleären Antikörpern (Anti-dsDNS-AK: 32 IU/ml, Norm: <10 IU/ml) wurde bei anfänglichem Verdacht auf eine Vaskulitis vor der stationären Zuweisung eine siebentägige Therapie mit 50 mg Prednisolon durchgeführt. Darunter waren die Rötung und die peripheren Ödeme im Bereich der Unterschenkel regredient bei gleichzeitiger Progredienz der nun zum Teil nekrotischen Ulzera (Abb. 1)
Abbildung 1: Läsionen am rechten Unterschenkel (A) und am linken medialen Unterschenkel (B) (Die Publikation erfolgt mit dem Einverständnis der Patientin.).
Befunde
Laborchemisch zeigte sich bei Eintritt eine normochrome, normozytäre Anämie mit einem Hämoglobinwert von 109 g/l (Norm: 120–160 g/l), eine Blutsenkungsgeschwindigkeit von 64 mm/h (Norm: <20 mm/h) und ein CRP von 33,9 mg/l (Norm: <5,0 mg/l). Neben einer erhöhten Laktatdehydrogenase (LDH) mit maximal 322 U/l (Norm: 135–214 U/l) waren die Anti-dsDNS-Antikörper mit 32 IU/ml (Norm: < 10 IU/ml) unverändert erhöht und das Komplement C3 mit 0,85 g/l (Norm: 0,90–1,80 g/l) leichtgradig erniedrigt. Die Proteinelektrophorese war vereinbar mit einer chronisch entzündlichen Reaktion und die Immunfixation ergab keinen Hinweis auf ein Paraprotein. Ein infektiologisches Screening für HIV sowie Hepatitis B und C fiel negativ aus. Ein Screening für EBV zeigte IgG-Antikörper bei fehlenden IgM-Antikörpern.
Die histologische Untersuchung der Biopsie aus einer der Läsionen an der rechten Wade ergab perivaskulär betonte Infiltrate von mittelgrossen bis grossen atypischen lymphoiden Zellen mit Infiltration und Zerstörung der Gefässwände. Immunhistochemische Analysen zeigten unter anderem eine Positivität für CD20 (ein Oberflächenprotein der B-Zellen). Fast in allen Tumorzellen konnte EBV nachgewiesen werden .
Diagnose
Anhand der Histologie und Immunhistologie wurde die Diagnose einer dermalen und subkutanen Infiltration eines angiozentrisch und angiodestruktiv wachsenden EBV-positiven DLBCL mit Ulzeration gestellt. In einer zytogenetischen Analyse mittels «fluorescence in situ hybridization» (FISH) konnten weder eine chromosomale Translokation und Aktivierung des «myelocytomatosis oncogene cellular homolog» (c-MYC), eines Protoonkogens, das die Expression von Zellwachstums- und Zelldifferenzierungsgenen reguliert, noch des «B-cell lymphoma-2» (Bcl-2), eines Gens, das den Zelltod (Apoptose) steuert, nachgewiesen werden. Eine genetische Veränderung in diesen oder verwandten Genen wird zur Prognoseabschätzung gebraucht. Insbesondere der Nachweis von zwei oder mehr genetischen Veränderungen gilt als prognostisch eher ungünstig.
Ein anschliessendes Staging zeigte in der Fluordesoxyglukose-Positronen-Emissions-Tomographie (FDG-PET) neben einer kräftigen FDG-Aktivität im Bereich beider Unterschenkel einen Uptake pulmonal links sowie im Bereich iliakaler Lymphknoten. Damit bestand der hochgradige Verdacht auf einen pulmonalen Befall, sodass die Diagnose eines EBV-positiven DLBCL im Stadium IV-B mit pulmonaler, nodaler und kutaner Manifestation gestellt wurde.
Therapie und weiterer Verlauf
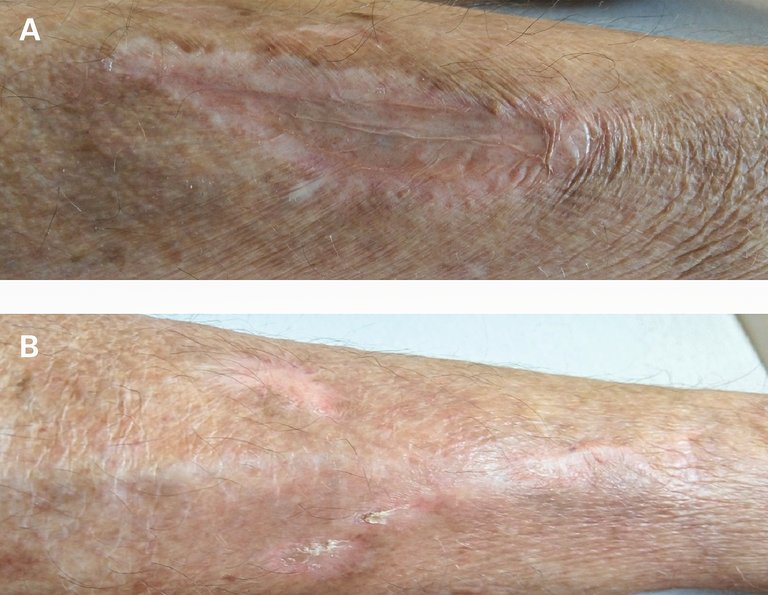
Es wurde eine Kombination einer Chemotherapie (CHOP [Cyclophosphamid, Hydroxydaunorubicin, Vincristin, Prednison]) mit dem Anti-CD20-Antikörper (Rituximab) begonnen, worunter die Ulzerationen im Verlauf abheilten (Abb. 2).
Abbildung 2: Läsionen am rechten (A) und linken medialen (B) Unterschenkel (Die Publikation erfolgt mit dem Einverständnis der Patientin.).
Bereits im kurzfristig, drei Monate nach Therapiebeginn, durchgeführten Verlaufs-FDG-PET zeigten sich die bekannten Lymphommanifestationen deutlich grössenregredient mit vermindert metabolischer Aktivität.
Diskussion
Zusammengefasst handelt es sich bei der Erkrankung dieser Patientin um ein EBV-positives DLBCL im Stadium IV-B mit pulmonalem, nodalem und kutanem Befall. Da der Befund in der Lunge nicht biopsiert wurde, ist eine Zweitneoplasie möglich, jedoch aufgrund des Verlaufes unwahrscheinlich. Klinisch stand der Hautbefall mit den Ulcera cruris im Vordergrund und führte zur Diagnosestellung.
Eine kutane Manifestation eines Lymphoms gehört zu den seltenen Differentialdiagnosen eines Ulcus cruris, dem am häufigsten eine vaskulär bedingte Problematik zugrunde liegt [1]. Häufige Ursachen sind auch Mikrozirkulationsstörungen bei Diabetes mellitus, meist kombiniert mit einer Neuropathie, physikalische Traumata, Infektionen oder autoimmunvermittelte Gefässentzündungen gehören zu den selteneren (Tab. 1) [1–3].
Wichtig für die Differentialdiagnose sind eine genaue Anamnese mit Beachtung des zeitlichen Verlaufs sowie des Ansprechens auf therapeutische Massnahmen, die Frage nach der Schmerzhaftigkeit sowie die klinische Untersuchung mit besonderem Augenmerk auf die Lokalisation, Wundbegrenzung, umgebende Haut, Atrophiezeichen sowie der Venen- und Pulsstatus.
An apparativer Diagnostik stehen die Duplexsonographie des Venensystems sowie die Erfassung des Knöchel-Arm-Index an erster Stelle. Bei atypischen Ulzera, die nach drei Monaten Therapie keine Rückbildungstendenz zeigen, rasch an Grösse zunehmen oder mit Begleiterscheinungen wie Livedo reticularis oder einer palpablen Purpura einhergehen, sollte eine fachärztliche dermatologische Überweisung und bei vaskulären Ulzera die frühzeitige Involvierung von Kolleginnen und Kollegen der Angiologie und Gefässchirurgie erfolgen. Unklare Befunde sollten insbesondere bei Verdacht auf eine systemische Erkrankung im Randbereich biopsiert und pathologisch oder dermatopathologisch beurteilt werden [1, 2].
Das DLBCL stellt eine sehr heterogene Erkrankung dar, in der nach histologischer, immunhistochemischer und genetischer Aufarbeitung verschiedene Subtypen unterschieden werden können [4]. Einer der Subtypen stellt das bei unserer Patienten diagnostizierte EBV-positive DLBCL/NOS (NOS: nicht weiter spezifiziert) dar, das in jedem Lebensalter auftreten und überlappende Eigenschaften mit einer lymphomatoiden Granulomatose aufweisen kann [4]. Eine extranodale Manifestation scheint bei diesem Subtyp häufig vorzukommen, wobei sich die Hautmanifestationen, insbesondere an den unteren Extremitäten, als erythematöse, typischerweise nicht ulzerierende Noduli oder Plaques präsentieren [4].
Abhängig vom Stadium der Erkrankung wird eine Chemoimmuntherapie mit oder ohne ergänzende Radiotherapie durchgeführt [4].
Die Prognoseeinschätzung bei dieser heterogenen Erkrankung hängt stark von verschiedenen klinischen und laborchemischen Parametern sowie der molekulargenetischen Einteilung ab, die im Internationalen Prognose Index (IPI) zusammengefasst werden [4].
Das Wichtigste für die Praxis
• Bei Ulcera cruris muss differentialdiagnostisch auch an neoplastische Erkrankungen gedacht werden. Eine zeitnahe Biopsie mit multiplen Entnahmen sollte bei atypischen Verläufen erwogen werden.
• Beim Epstein-Barr-Virus-(EBV-)positiven diffusen, grosszelligen B-Zell-Lymphom (DLBCL) handelt es sich um ein aggressives Non-Hodgkin-Lymphom.
• Eine histologische Diagnosesicherung mit Durchführung einer immunhistochemischen und zytogenetischen Analyse ist die Voraussetzung für eine Prognoseeinschätzung und optimale Therapie.
Disclosure statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Korrespondenz
Dr. med. Joscha von Rappard Klinik für Innere Medizin und Nephrologie Hirslanden Klinik St. Anna Sankt-Anna-Strasse 32 CH-6006 Luzern joscha.vonrappard[at] hirslanden.ch
Literatur
1 Meyer V, Kerk N, Meyer S, Goerge T. Differential diagnosis and therapy of leg ulcers. J Dtsch Dermatol Ges. 2011;9(12):1035–51.
2 Sarkar PK, Ballantyne S. Management of leg ulcers. Postgrad Med J. 2000;76(901):674–82.
3 Miller A, Ruzicka T. Differentialdiagnose des Ulcus cruris. Hautarzt. 2001;52(7):593–603.
4 Castillo JJ, Beltran BE, Miranda RN, Young KH, Chavez JC, Sotomayor EM. EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2018 update on diagnosis, risk-stratification and management. Am J Hematol. 2018;93(7):953–62.
5 Dissemond J. Chronisches Ulcus cruris. Hautarzt. 2017;68(8):614–20.