Die notfallmässige hausärztliche Zuweisung einer 61-jährigen Patientin erfolgte zur Weiterabklärung einer persistierenden Kreatinkinase-Erhöhung trotz Sistieren einer Statintherapie.
Hintergrund
Statine sind effiziente Medikamente zur Primär- und Sekundärprävention kardiovaskulärer Ereignisse. Muskeltoxizität ist eine klinisch bedeutende unerwünschte Wirkung der Statine. Diese kann von Myalgien mit einer milden Erhöhung der Kreatinkinase (CK) bis hin zu Myotoxizität einschliesslich Rhabdomyolyse variieren. Selten tritt eine immunvermittelte nekrotisierende Myopathie («immune-mediated necrotising myopathy» [IMNM]) auf. Die IMNM ist eine Autoimmunerkrankung, die durch proximale Muskelschwäche, erhöhte CK-Werte im Serum, ein myopathisches Muster in der Elektromyographie sowie durch Nekrose und Entzündung in der Muskelbiopsie und durch einen Anstieg der Anti-3-Hydroxy-3-Methylglutaryl-Coenzym-A-Reduktase-(HMGCR-)Antikörper gekennzeichnet ist. Im Folgenden möchten wir einen entsprechenden Fall präsentieren.
Fallbericht
Anamnese
Die notfallmässige hausärztliche Zuweisung der 61-Jährigen Patientin erfolgte zur Weiterabklärung einer persistierenden CK-Erhöhung trotz Sistieren einer Statintherapie.
Die Patientin berichtete über eine seit vier Wochen bestehende Schwäche der Arme und Beine, diese würden sich «wie Blei» anfühlen. Sie konnte daher kaum mehr gehen, Treppensteigen verursachte grosse Schwierigkeiten. Durch den Hausarzt wurde die bestehende Therapie mit Atorvastatin bei einer CK von knapp 10 000 U/l gestoppt. In der Folge kam es initial zu einer leichten Besserung der Beschwerden mit jedoch persistierend deutlich erhöhter CK, sodass die Patientin bei unzureichender klinischer Besserung zur Weiterabklärung zu uns überwiesen wurde. Fieber, Infektsymptome, pulmonale Probleme, Myalgien, Visusprobleme, ein Raynaud-Syndrom oder eine Sicca-Symptomatik verneinte die Patientin.
Aus der Anamnese bestand als Nebendiagnose eine Hypercholesterinämie, die bis Dezember 2019 mit Atorvastatin 40 mg (seit zwei Jahren) therapiert wurde.
Status und Befunde
Bei Eintritt präsentierte sich eine 61-jährige Patientin in ordentlichem Allgemeinzustand und kreislaufstabil. In der klinischen Untersuchung fanden sich bis auf eine deutliche Hüftbeugerschwäche beidseits keine Auffälligkeiten.
Im Labor zeigten sich eine deutliche erhöhte CK (7405 U/l), eine Aspartat-Aminotransferase (ASAT) von 312 U/l, eine Alanin-Aminotransferase (ALAT) von 423 U/l sowie eine Laktatdehydrogenase (LDH) von 1855 U/l, die übrigen Werten lagen im Normbereich. Zudem waren die antinukleären Antikörper und myositisspezifischen Antikörper negativ.
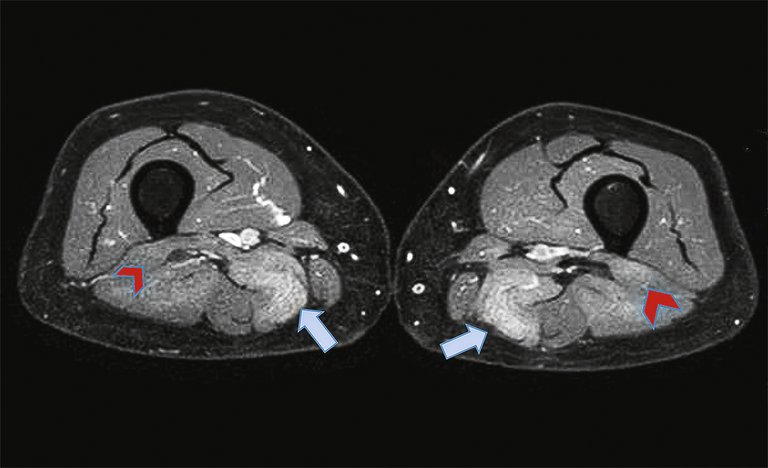
Eine Magnetresonanztomographie (MRT) der Oberschenkel zeigte eine symmetrisch ausgebildete Myositis der Muskulatur des Beckengürtels sowie des posterioren Oberschenkelkompartimentes mit symmetrisch ödematösen Veränderungen mit entsprechender Kontrastmittelaufnahme, die beidseits die Hüftabduktoren, gering den Musculus (M.) gluteus maximus, Muskeln der Adduktorenloge und den M. obturator externus sowie den M. iliopsoas betraf; weiter distal waren in erster Linie der M. semimembranosus und der kurze Kopf des M. biceps femoris betroffen. (Abb. 1)
Abbildung 1: Magnetresonanztomogramm, T1-Sequenz nach Gadolinium mit Fettsuppression: Hier sind die symmetrischen ödematösen Veränderungen mit entsprechender Kontrastmittelaufnahme zu sehen. Das posteriore Oberschenkelkompartiment ist besonders betroffen: Musculus (M.) semimenbranosus (blau) und der kurze Kopf der M. biceps femoris (rot).
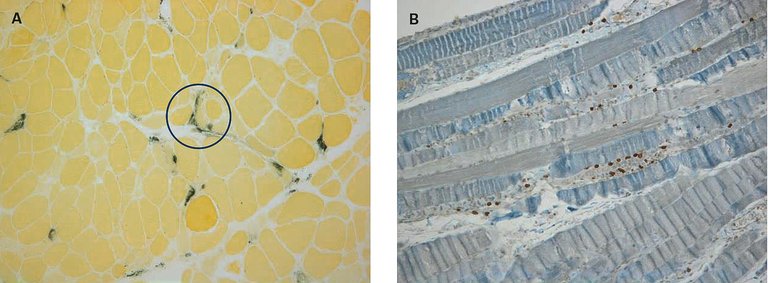
Es erfolgte eine Biospie aus dem M. gluteus medius rechts, die eine Skelettmuskulatur mit zahlreichen nekrotischen und regenerierenden Fasern mit schütterem Entzündungsinfiltrat und vermehrter perimysialer Ablagerung von alkalischer Phosphatase, entsprechend einer nekrotisierenden Myopathie (Abb. 2A) zeigte. Immunhistochemisch bestand das entzündliche Infiltrat vor allem aus CD8-positiven T- Lymphozyten, weniger aus CD4-positiven T-Lymphozyten ohne ersichtliche CD20-positive B-Lymphozyten. CD68-positive Makrophagen zeigten die Myophagozytosen an, ebenso fanden sich endomysial einige Makrophagen (Abb. 2B).
Abbildung 2:A) Perimysiale Ablagerung von alkalischer Phosphatase (Färbung: AP = alkalische Phosphatase; Vergrösserung 100×). B)Entzündliches Infiltrat vor allem aus CD8-positiven T-Lymphozyten (Färbung: CD8 = Marker für zytotoxische T-Lymphozyten; Vergrösserung 100×).
Dieser Befund war am besten mit einer IMNM vereinbar. Eine Einschlusskörperchenmyositis war bei Fehlen von hypertrophen Fasern, mitochondrialer Pathologie und veränderten Vakuolen unwahrscheinlich. Für eine Dermatomyositis war der Befund bei fehlender perifaszikulärer Atrophie und fehlenden Komplementablagerungen ebenfalls nicht typisch.
Die HMGCR-Antikörper waren mit >200 U/ml stark erhöht, sodass die Diagnose einer statininduzierten immunvermittelten nekrotisierenden Myopathie gestellt werden konnte.
Therapie und Verlauf
Nach Biopsie wurde eine systemische Glukokortikoidtherapie mit Prednisolon 50 mg (1 mg/kg) begonnen (Abb. 3). Zusätzlich wurde dann eine Basistherapie mit Methotrexat (Februar 2020, initial 15 mg/Woche, dann im Verlauf Steigerung auf 20 mg/Woche) eingeleitet. Vier Wochen nach Beginn der Glukokortikoidtherapie, im Februar 2020, und fünf Monate nach Beginn der Glukokortikoidtherapie, im Juni 2020, wurden bei persistierender Schwäche auch intravenöse Immunglobuline (IVIg) verabreicht, dies in einer Gesamtdosis von 2 g/kg Körpergewicht (KG) verteilt über fünf Tage.
Im Verlauf konnte ein Rückgang der CK (3891 U/l nach zwei Wochen, im Juni 2020 1714 U/l) dokumentiert werden (Abb. 3).
Abbildung 3: Schematische Darstellung des Kreatinkinase-(CK-) und Therapieverlaufs.
Die Behandlung mit Glukokortikoiden und Methotrexat wurde insgesamt gut toleriert, aber es zeigte sich bis etwa Mitte März 2020 (2,5 Monate nach Beginn der Therapie) noch eine deutliche subjektive und objektivierbare Schwäche der proximalen Oberschenkelmuskulatur ohne Schmerzen. Ab etwa Ende März 2020 verspürte die Patientin eine deutliche Verbesserung der Symptomatik, sie konnte problemlos bis zu vier Kilometern laufen, und auch Treppensteigen und längere E-Bike-Touren waren kein Problem mehr. Bei der letzten Verlaufskontrolle im September 2020 (insgesamt neun Monate nach Beginn der Therapie) war die Patientin praktisch beschwerdefrei.
Diskussion
Statine sind dafür bekannt, toxische Myopathien zu verursachen. Die statinassozierte Muskeltoxizität (SAMT) stellt die am häufigsten gemeldete unerwünschte Statinnebenwirkung dar und macht etwa zwei Drittel aller unerwünschten Ereignisse aus. Die häufigsten Muskelsymptome sind Schmerzen, Schweregefühl, Steifheit und Krämpfe mit oder ohne subjektive Schwäche [1]. Am häufigsten sind Symptome im Bereich der Beinmuskulatur (Oberschenkel, Waden), es wurden aber auch Rücken-, Nacken-, Schulter- und generalisierte Muskelsymptome beschrieben [1]. Auch über mit Sehnenentzündung assoziierte Schmerzen wurde berichtet [1]. Ungefähr 40% der Patientinnen und Patienten mit SAMT bemerken einen möglichen Auslöser: am häufigsten eine ungewöhnliche körperliche Anstrengung oder ein neues Medikament [1]. Muskelschmerzen treten bei drei Viertel der SAMT-Patientinnen und -Patienten zeitweise auf und sind bei einem Viertel konstant [1].
Die SAMT tritt am häufigsten im ersten Behandlungsjahr auf mit einer mittleren Zeit bis zum Einsetzen von einem Monat. Über 80% der Patientinnen und Patienten berichten, dass sie vor der Behandlung mit Statinen keine ähnlichen Symptome hatten [1]. Die Muskelsymptome sind in den meisten SAMT-Fällen (~70–80%) ausreichend intensiv, um die täglichen Aktivitäten zu beeinträchtigen [1]. Die selteneren schweren Myopathien und Rhabdomyolysen können direkt zu einem Spitalaufenthalt führen.
Die SAMT ist in der Präsentation heterogen, sodass die Falldefinitionen zwischen den Studien variieren. Kürzlich wurde daher die Nomenklatur standardisiert und die SAMT in sieben verschiedene phänotypische Kategorien eingeteilt [2] (Tab. 1).
Tabelle 1: Die Klassifizierung und die geschätzten Häufigkeiten basieren auf Alfirevic et al. [2], die Häufigkeit der Myalgie auf Parker et al. [3].
Klinik und Laborparameter
Inzidenz
SAMT 0
Asymptomatische Erhöhungen der CK <4× der Obergrenze des Normalwerts (ULN)
1,5–26% der Patienten
SAMT 1–2
Häufige Myalgien (Schmerzen, Krämpfe und/oder Schwäche) ohne (SAMT 1) oder geringfügige CK-Erhöhungen (<4× ULN, SAMT 2)
Etwa 5% der Patienten
SAMT 3
Zunehmend seltene Myopathie mit CK >4× aber <10× ULN
5–100 000 Patienten/Jahr
SAMT 4
Schwere Myopathie mit CK >10× aber <50× ULN
0,11% der Patienten
SAMT 5
Seltene, aber möglicherweise lebensbedrohliche Rhabdomyolyse mit entweder CK >10× ULN, Muskelsymptomen und Nierenfunktionsstörungen oder CK >50× ULN
0,1–8,4/100 000 Patienten/Jahr
SAMT 6
Sehr seltene anti-HMGCR-positive immunvermittelte nekrotisierende Myopathie
Die IMNM ist eine kürzlich anerkannte Kategorie der entzündlichen Myopathien. Die Autoimmunität der IMNM wird durch die häufige Assoziation mit zwei spezifischen Autoantikörpern belegt, nämlich Antikörper gegen HMGCR und «signal-recognition particle» (SRP). Bei Patientinnen und Patienten, die Statine einnehmen, beträgt die geschätzte IMNM-Inzidenzrate 2–3 pro 100 000 Patientinnen und Patienten [2], wobei das Risiko bei Personen über 50 Jahre erhöht ist [3].
Die betroffenen Patientinnen und Patienten sind in der Regel Erwachsene mit einem Durchschnittsalter von etwa 59 Jahren, tendenziell weiblich. Bei Kindern ist die Entwicklung einer statininduzierten IMNM sehr selten. In den letzten Jahren wurden aber mehrere pädiatrische Patientinnen und Patienten mit Anti-HMGCR-Myopathie identifiziert ohne vorherige Exposition gegenüber Statinen.
Das klinische Hauptmerkmal einer IMNM ist eine symmetrische, proximale Muskelschwäche an Armen und Beinen (Tab. 2). Die Krankheitsaktivität korreliert mit dem Ausmass der individuell messbaren und objektivierbaren Muskelkraft. Die klinischen Symptome können akut (innert Tagen bis Wochen) oder subakut (innert Monaten) auftreten. Im Labor kann ein massiver Anstieg der CK nachgewiesen werden [4], diese ist aber als Verlaufsparameter nur bedingt geeignet, da eine Korrelation mit der Muskelkraft im Rahmen der IMNM noch nicht nachgewiesen werden konnte [5].
Tabelle 2: Schematische Darstellung der «immune mediated necrotising myopathy»-(IMNM-)Merkmale.
In der Elektromyographie von Betroffenen kann man spontane elektrische Entladungen (Fibrillationspotential) und steil ansteigende Wellen nachweisen. Radiologisch ist im MRT ein Ödem der betroffenen Muskulatur, im Sinne einer aktiven Inflammation, zu beobachten. In der Biopsie zeigt sich das Bild einer nekrotisierende Myopathie ohne Vaskulitis.
Der pathopysiologische Mechanismus der statininduzierten IMNM ist aktuell immer noch nicht sicher geklärt.
Die HMGCR ist ein membranassoziiertes Protein, das überwiegend im endoplasmatischen Retikulum lokalisiert ist und in geringerem Masse auch in Peroxisomen vorhanden sein kann. Sie ist in die Cholesterinbiosynthese involviert und wird durch Statine inhibiert.
Es wurde gezeigt, dass eine Statinexposition zu einer Überexpression von HMGCR in mehreren Geweben und Zelltypen führt. Varianten von HMGCR oder kryptische Epitope und/oder Polymorphismen im HLA-Molekül, die durch Medikamente demaskiert sind, können die T-Zell-Rezeptoren-Aktivierung erhöhen und die Autoimmunität initiieren; eine immunvermittelte Verletzung des Muskels ist dann gegen HMGCR oder andere Antigene gerichtet, die von einem Anti-HMGCR-Antikörper erkannt werden. Die Reparaturversuche von Muskelfasern führen zu einer erhöhten HMGCR-Expression und zu einer anhaltenden Autoimmunität, anschliessend mit Atrophie und Degeneration von Muskelfasern.
Diskutiert wird, ob der immunogenetische Hintergrund der Patientin / des Patienten auch eine Rolle bei der Prädisposition zur Entwicklung einer IMNM spielen kann. Das Vorhandensein des HLA-DRB1*11: 01-Allels ist ein gut etablierter immunogenetischer Risikofaktor für die Entwicklung dieser Krankheit (Erwachsene zeigen häufig das humane Leukozytenantigen [HLA] DRB1*11:01 und Kinder DRB1*07:01) [6, 7]. Ursprünglich bei fast 70% der Patientinnen und Patienten identifiziert, wurde es nun in mehreren Kohorten mit unterschiedlicher ethnischer Herkunft als immunogenetischer Risikofaktor validiert.
Aktuell existieren noch keine internationalen Leitlinien zur Therapie der IMNM [8, 9]. Das Absetzen der Statintherapie ist in der Regel der erste und wichtigste Schritt bei der Behandlung.
Die immunsuppressive Therapie ist jedoch der Eckpfeiler der Behandlung. Selva-O’Callaghan et al. [10] berichten, dass IVIg die beste Option zur Behandlung einer statininduzierten IMNM zu sein scheinen, da es das einzige Mittel ist, das sich zur Behandlung von entzündlichen Myopathien als wirksam erwiesen hat. Nichols et al. [11] diskutieren andererseits die Verwendung von Immunoglobulinen nur in Fällen, die auf eine immunsuppressive Therapie nicht ansprechen oder in Fällen, die bei der Diagnosestellung besonders schwerwiegend sind. Andere Immunsuppressiva wie Azathioprin und Methotrexat wurden ebenfalls mit guter Wirkung verwendet, ebenso seltener Calcineurin-Inhibitoren und Mycophenolatmofetil [10, 11]. Es wurde beobachtet, dass viele Anti-HMGCR-positive Patientinnen und -Patienten initial nach Prednisongabe nur eine geringe Besserung zeigen, und deshalb wird eine Kombinationstherapie mit in der Regel zwei Immunsuppressiva benötigt, bis eine Verbesserung der Muskelkraft und eine Normalisierung der CK-Werte nachgewiesen werden kann [5–9].
Obwohl kein klarer Zusammenhang zwischen spezifischen Statinen und der Entwicklung von IMNM gezeigt wurde, haben frühere Berichte eine grössere Anfälligkeit von Patientinnen und Patienten, die Atorvastatin oder Simvastatin einnehmen, ergeben [11].
Die Prognose ist in der Regel gut, obwohl bei einigen Patientinnen und Patienten Rückfälle auftreten können und eine Langzeittherapie erforderlich sein kann.
Das Wichtigste für die Praxis
• Die statininduzierte immunvermittelte nekrotisierende Myopathie (IMNM) ist eine seltene Komplikation einer Statintherapie.
• Die Erkrankung ist klinisch gekennzeichnet durch eine proximale Schwäche der Extremitätenmuskulatur, im Labor besteht eine persistierende Erhöhung der Kreatinkinase trotz Sistieren der Statintherapie und eines Nachweises von Anti-HMGCR-Antikörpern.
• Die Erkrankung muss mittels immunsuppressiver Therapie (Glukokortikoide, Methotrexat oder Azathiorin, eventuell intravenöse Immunoglobuline) behandelt werden.
Verdankung
Wir danken Herrn PD Dr. med. C. Schäffeler, Radiologie, Kantonsspital Chur, für die Bereitstellung und Befundung der MRT-Bilder und Herrn Dr. med. K. Frontzek, Neuropathologie, Universitätsspital Zürich, für die Histologiebilder.
Disclosure statement
Die Autoren haben deklariert, keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag zu haben.
Korrespondenz
Dr. med. Ofelia Boiocchi Departement Innere Medizin Kantonsspital Graubünden Loëstrasse 170 CH-7000 Chur Oboiocchi[at]hotmail.com
Literatur
Die vollständige Literaturliste finden Sie in der Online-Version des
1 Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—The primo study. Cardiovasc. Drugs Ther. 2005;19:403–14.
2 Alfirevic A, Neely D, Armitage J, Chinoy H, Cooper RG, Laaksonen R, Carr DF, Bloch KM, Fahy J, Hanson A, et al. Phenotype standardization for statin-induced myotoxicity. Clin. Pharmacol. Ther. 2014;96.
3 Parker BA, Capizzi JA, Grimaldi AS, Clarkson PM, Cole SM, Keadle J, Chipkin S,Pescatello LS, Simpson K, White CM, et al. Effect of statins on skeletal muscle function. Circulation. 2013;127:96–103.
4 Kishi T, Rider LG, Pak K, et al. Childhood Myositis Heterogeneity Study Group. Association of anti-3-hydroxy-3-methylglutaryl-coenzyme a reductase autoantibodies with DRB1*07:01 and severe myositis in juvenile myositis patients. Arthritis Care Res. 2017;69:1088–94.
5 Laccarino L, et al. Assessment of patients with idiopathic inflammatory myopathies and isolated creatin-kinase. Auto Immun Highlights. 2014;5(3):87–94.
6 Mammen AL, Gaudet D, Brisson D, et al. Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res. 2012;64:1233–7.
7 Allenbach Y et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine. 2014;93:150–7.
8 Meyer A, Troyanov Y, Drouin J, et al. Statin-induced anti-HMGCR myopathy: successful therapeutic strategies for corticosteroid-free remission in 55 patients. A. Arthritis Res Ther. 2020;22(1):5.
9 Saleh Y, Herzallah K, Hassanein M, Chang HT. Statin- induced necrotizing autoimmune myopathy: an uncommon complication of a commonly used medication. J Saudi Heart Assoc. 2019;31(4):269–72.
10 Selva-O’Callaghan A, Alvarado-Cardenas M, Pinal-Fernandez I, et al. Statin-induced myalgia and myositis: An update on pathogenesis and clinical recommendations. Expert Rev Clin Immunol. 2018;14:215–24.
11 Nichols L, Pfeifer K, Mammen AL, Shahnoor N, Konersman CG. An unusual case of statin-induced myopathy: Anti-HMGCoA Necrotizing Autoimmune Myopathy. J Gen Intern Med. 2015;30:1879–83.