Ein sonst gesunder 43-Jähriger leidet seit 1½ Jahren unter rezidivierenden Episoden allgemeiner Schwäche und Leistungsintoleranz, mit Hypotonie, Tachykardie, generalisierten Ödemen, Schwindel und diffusen Muskel- und Gliederschmerzen.
Hintergrund
Wir beschreiben den Fall eines Patienten mit den typischen Symptomen und Befunden eines systemischen «capillary leak»-Syndroms («Clarkson’s disease»). Der Nachweis einer monoklonalen Gammopathie, die typischerweise mit diesem seltenen und wenig bekannten Syndrom assoziiert ist, lieferte nach einer langen Leidensgeschichte des Patienten den Schlüssel zur Diagnose. Unter einer hochdosierten intravenösen Gammaglobulintherapie ist die Erkrankung seit drei Jahren in Remission.
Fallbericht
Anamnese
Ein sonst gesunder, 43-jähriger Patient leidet seit 1½ Jahren unter rezidivierenden Episoden von allgemeiner Schwäche und Leistungsintoleranz, verbunden mit Hypotonie, Tachykardie, generalisierten Ödemen, Schwindel und diffusen Muskel- und Gliederschmerzen. Diese Episoden dauern 48–72 Stunden und wiederholen sich alle 5–6 Tage. Sie gehen einher mit einer reduzierten Miktionsfrequenz und einer sich anschliessenden polyurischen Phase. Der Patient ist während der Episoden nicht in der Lage, seiner Arbeitstätigkeit nachzugehen. Seine sportliche Aktivität muss er aufgrund der Symptome aufgeben. Er wird kardiologisch und neurologisch inklusive Elektromyographie abgeklärt, mit unauffälligen Befunden. Die Blutuntersuchung zeigt eine leichtgradige Anämie mit einem Hämoglobinwert von 118 g/l. Im Liquor finden sich nebenbefundlich monoklonale Banden, als Ausdruck der dann in der Immunfixation im Serum diagnostizierten geringen systemischen Paraproteinämie vom Typ IgG lambda (nicht quantifizierbar). Der Patient wird deshalb zur hämatologischen Abklärung zugewiesen.
Status und Befunde
Bei der Erstkonsultation ist der Patient beschwerdefrei, normoton und normokard, sein Gewicht beträgt 75 kg bei einer Grösse von 182 cm. Es bestätigt sich eine leichtgradige normochrome, normozytäre Anämie mit einem Hämoglobinwert von 128 g/l und einem Hämatokrit von 37,2%. Der Albuminwert beträgt 42 g/l.
Zehn Tage später erfolgt eine Knochenmarkdiagnostik zur weiteren Abklärung der monoklonalen Gammopathie unklarer Signifikanz (MGUS) und der leichtgradigen Anämie. Zu diesem Zeitpunkt fühlt sich der Patient sehr geschwächt, er hat Schwindel, diffuse Muskelschmerzen und periphere Ödeme. Das Gewicht ist auf 79 kg angestiegen, es besteht eine Sinustachykardie bei normalem Blutdruck. Der Hämoglobinwert beträgt 194 g/l, der Hämatokritwert 56,9%.
Die Resultate der Knochenmarkuntersuchung sind unauffällig, insbesondere lässt sich keine Infiltration durch ein Multiples Myelom oder ein Non-Hodgkin-Lymphom finden. Die Immunphänotypisierung zeigt keine klonale Plasmazellpopulation. Bei normaler Zellularität und Morphologie der Hämatopoese finden sich keine Hinweise auf eine myeloproliferative Erkrankung. Eine JAK2-Mutationsanalyse ist negativ. Die Paraproteinämie vom Typ IgG lambda wird bestätigt, die Ratio der freien Leichtketten kappa/lambda ist normal. Eine Albuminurie liegt nicht vor. Das native «low dose» Ganzkörper-CT ist normal.
In der Folge können bei weiteren Schwächeepisoden mit Hypotonie und Tachykardie wiederholt eine Hämokonzentration mit Anstieg des Hämatokritwertes von 36 bis auf 62% und des Hämoglobinwertes von 128 bis auf 214 g/l nachgewiesen werden. Der Hämatokritanstieg ist verbunden mit einer Gewichtszunahme von bis zu 5 kg und einem Absinken des Albumins bis auf 30 g/l (Laborwerte siehe Tab. 1).
Tabelle 1: Laborwerte bei Erstkonsultation und bei einer «capillary leak»-Episode.
Wert
Erstkonsultation
Episode
Hämoglobin (g/l)
128
214
Hämatokrit (%)
37,2
61,8
Leukozyten (G/l)
5,06
16,28
Neutrophile (G/l)
3,46
12,98
Albumin (g/l)
42
30
Kreatinin (mmol/l)
80
170
Diagnose
Aufgrund der rezidivierenden Episoden mit Hypotonie, Hämatokriterhöhung, peripheren Ödemen und Hypoalbuminämie ohne Albuminurie und dem Nachweis eines Paraproteins stellen wir die Diagnose eines Monoklonale-Gammopathie-assoziierten systemischen «capillary leak»-Syndroms («Clarkson’s Disease»).
Therapie und Verlauf
Gemäss Empfehlungen der «EurêClark Study Group» beginnen wir eine hochdosierte intravenöse Gammaglobulintherapie (2 mg/kg Körpergewicht alle vier Wochen). Unter dieser Behandlung sistieren die «capillary leak»-Episoden vollständig mit Verschwinden der Symptome und der Gewichtsschwankungen (Abb. 1) sowie Sistierung des Hämatokritanstiegs (Abb. 2) und Albuminabfalls.
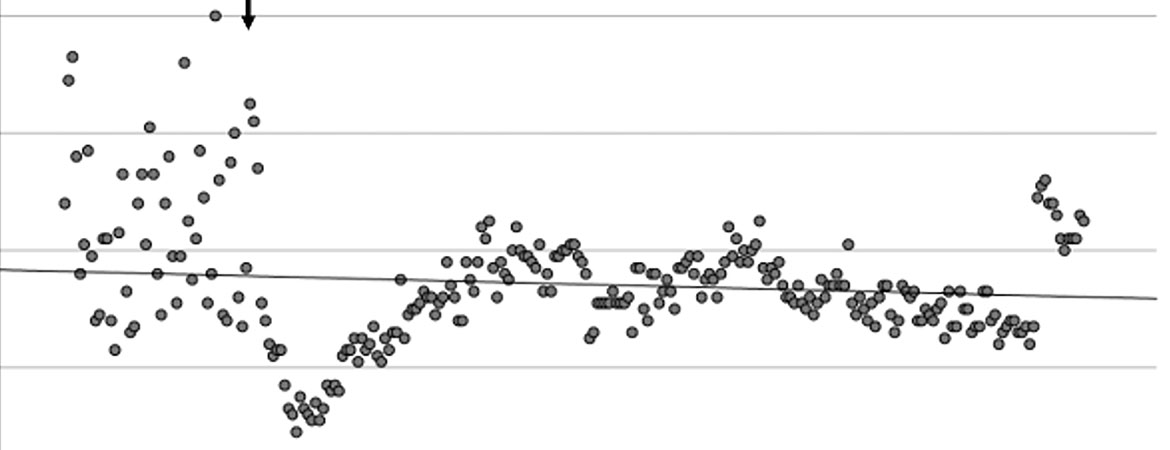
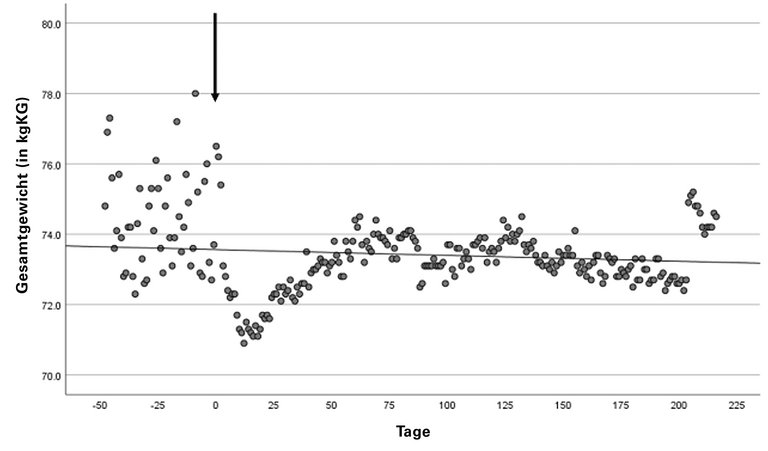
Abbildung 1: Messung des Patientengewichts 49 Tage vor bis 216 Tage nach Beginn der Therapie. Der Pfeil markiert den Zeitpunkt des Therapiebeginns. Es zeigt sich eine deutliche Reduktion des Körpergewichts (prätherapeutisch vs. Therapie: 74,36 kg ± 1,39 kg vs. 73,22 kg ± 0,85 kg; Mittelwert ± SD). kg: Kilogramm; KG: Körpergewicht.
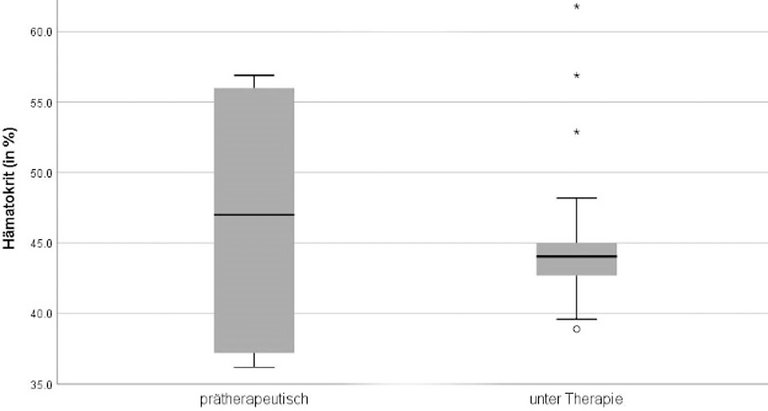
Abbildung 2: Hämatokritwerte vor und während der Therapie (46,7% ± 10,2 vs. 45,3% ± 5,4; Mittelwert ± SD).
Es kommt seit Therapiebeginn zu keinen Arbeitsausfällen mehr und der Patient erreicht wieder seine volle körperliche Leistungsfähigkeit. Der Versuch, nach einem Jahr die Immunglobulindosierung auf 1 g/kg Körpergewicht alle vier Wochen zu reduzieren, führt zu einer erneuten «capillary leak»-Episode, weshalb die Behandlung mit 2 g/kg weitergeführt wird. Auch eine Verlängerung des Dosisintervalls hat ein Rezidiv zur Folge. Drei Jahre nach Therapiebeginn besteht weiterhin eine Remission.
Diskussion
Das systemische «capillary leak»-Syndrom wurde erstmals 1960 von B. Clarkson beschrieben [1]. Er publizierte den Fall einer 34-jährigen Frau, die «periodisch und unerklärlich an einem plötzlichen Verlust von Plasma aus ihrem Gefässbett litt und bei der sich eine Hypovolämie und Schock entwickelte». Die Erkrankung war charakterisiert durch wiederholte Episoden von erhöhter Kapillarpermeabilität mit Ödemen, Hämokonzentration, Hypalbuminämie und Hypotonie und es wurde ein abnormes Protein in der Gammaglobulinfraktion nachgewiesen. Schliesslich verstarb die Patientin an einem hypovolämen Schock.
2010 wurde eine Serie von 25 Patienten aus der Mayo-Klinik mit systemischem «capillary leak» publiziert[2]. Die Diagnose basierte auf einer eindeutigen Dokumentation von wiederholten Attacken von Hypotension, erhöhter Hämatokritkonzentration, peripheren Ödemen und Hypalbuminämie ohne Albuminurie. Eine monklonale Gammopathie wurde nicht als obligates Kriterium betrachtet, jedoch bei 19 der 25 Patienten nachgewiesen. Die meisten Patienten hatten Prodromalsymptome wie Müdigkeit und Schwindel, zum Teil auch Myalgien und Halsschmerzen sowie eine reduzierte Miktionsfrequenz. Während «capillary leak»-Attacken kam es zu einem medianen Anstieg des Hämatokrites um 19,8% (14,8–24) auf 60,5% (53,6–65,6). Der mediane Albuminwert sank während einer Attacke um 21 g/l (18–25) und der mediane Kreatininwert stieg um 132 µmol/l (88–200). 36% der Patienten entwickelten eine Rhabdomyolyse, 20% ein Kompartmentsyndrom, das zum Teil eine Fasziotomie erforderte. Die Paraproteinkonzentration war typischerweise gering (median 6 g/l). Die Progressionsrate in ein Multiples Myelom betrug 0,7% per Personenjahr und war damit vergleichbar mit derjenigen eines MGUS.
Die «European Clarkson Disease (EurêClark) Registry» ist eine international Studiengruppe, die Daten von Patienten mit Monoklonale-Gammopathie-assoziiertem systemischem «capillary leak»-Syndrom sammelt. Die Gruppe veröffentlichte 2017 im American Journal of Medicine die Therapieergebnisse einer Kohorte von 69 Patienten nach regelmässiger Verabreichung von intravenösem Immunglobulin zur Rezidivprophylaxe. Die Kriterien für die Diagnose einer «Clarkson’s disease» waren: 1. Vorhandensein einer monoklonalen Gammopathie; 2. eine oder mehrere Episoden akuter Hypovolämie und interstitieller Ödeme; 3. Hämokonzentration mit paradoxer Hypoproteinämie; 4. Ausschluss einer anderen Ursache eines sekundären «capillary leak»-Syndroms oder einer Hypoproteinämie.
Die Kohorte bestand aus 35 Frauen und 34 Männern, das mediane Alter bei Erstmanifestation lag bei 52 Jahren (±12), bei Diagnose bei 53,5 Jahre (±12). Alle hatten eine Gammopathie vom Typ IgG mit einer medianen Paraproteinkonzentration von 4,4 g/l (2–8).
Zur Prävention von «capillary leak»-Episoden erhielten die Patienten vor dem Jahr 2000 Theophyllin und Terbutalin, danach wurde intravenöses Immunglobulin vermehrt als Erstlinientherapie eingesetzt.
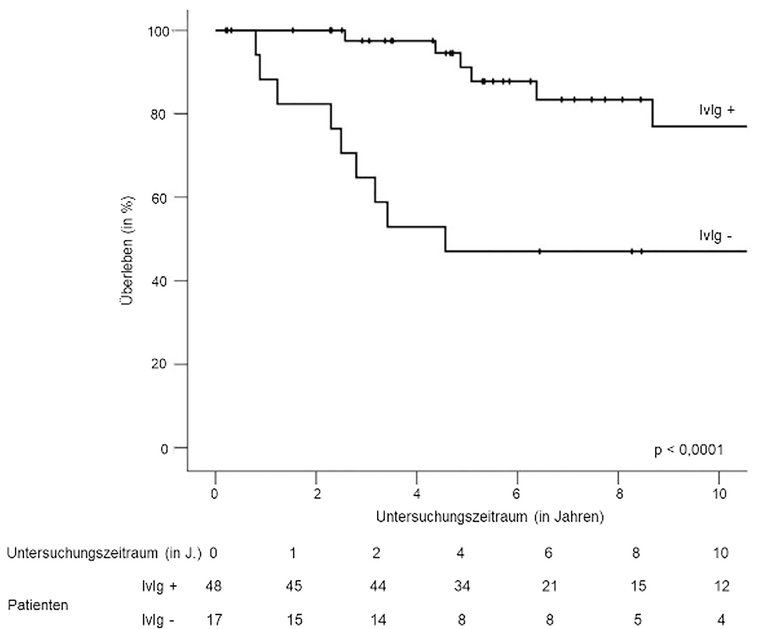
Bei einem medianen Follow-up von 5,1 Jahren (2,5–9,7) entwickelten fünf Patienten ein Multiples Myelom. 24 starben nach einer medianen Zeit von 3,3 Jahren (0,9–8): 20 infolge einer schweren Attacke, vier an einem multiplen Myelom. Patienten, die mit intravenösem Immunglobulin behandelt wurden, hatten signifikant weniger schwere Rückfallattacken als solche, die kein intravenöses Immunglobulin erhielten (45,8% vs. 94,1%) und das 5- respektive 10-Jahres-Überleben war signifikant besser (91 vs. 47% respektive 77 vs. 37%) (Abb. 3 [3]).
Die Pathogenese des systemischen «capillary leak»-Syndroms und die ätiologische Bedeutung des Paraproteins ist weiterhin unklar. Wahrscheinlich sind humorale Faktoren, die eine reversible Dysfunktion der mikrovaskulären Endothelzellbarriere zur Folge haben, verantwortlich. So wurden während Attacken erhöhte Werte bekannter angiogener Permeabilitätsfaktoren wie «vascular endothelial growth factor» (VEGF) und Angiopoietin 2 sowie von Interleukinen und anderen Entzündungsfaktoren gefunden. Strukturelle Gefässveränderungen konnten nicht nachgewiesen werden [4].
Die Diagnose eines systemischen «capillary leak»-Syndroms bleibt eine klinische Diagnose mit rezidivierenden Episoden von akuter Hypovolämie und interstitiellen Ödemen, Hämokonzentration, Hypalbuminämie ohne Albuminurie, typischerweise Nachweis eines Paraproteins und Ausschluss einer anderen Ursache wie Sepsis, Anaphylaxie oder Angioödem. Die Episoden sind selbstlimitierend, dauern wenige Tage und sind in Häufigkeit und Schwere stark variabel. Eine schwere Episode kann zu einem lebensbedrohlichen Schockzustand mit akuter Niereninsuffizienz und Multiorganversagen führen. Thrombosen und Lungenembolien als Folge der Hämokonzentration, Perkarderguss mit Perikardtamponade und Kompartmentsyndrom der Extremitäten mit Rhabdomyolyse durch die peripheren Ödeme können vorkommen.
Therapeutisch ist in der akuten Phase in erster Linie eine supportive Behandlung notwendig, mit Korrektur der Hypovolämie durch Infusion kristalloider Lösungen und eventuell Albuminsubstitution. Bei schweren Verläufen können eine intensivmedizinische Betreuung und der Einsatz von Vasoaktiva erforderlich sein, bei thrombotischen Komplikationen eine Antikoagulation. In der Erholungsphase des «capillary leak» besteht die Gefahr einer Flüssigkeitsüberladung mit Lungenödem und es kann eine intensive diuretische Therapie notwendig sein. Beschrieben sind Fälle, bei denen in der Annahme einer Polyzythämia vera irrtümlich Aderlässe durchgeführt wurden [5].
Zur Verhinderung von erneuten «capillary leak»-Episoden wird aufgrund oben erwähnter Analyse der «European Clarkson Disease (EurêClark) Registry» der Einsatz von hochdosiertem Immunglobulin, initial in einer Dosierung von 2 g/kg Körpergewicht vierwöchentlich, empfohlen. Damit scheinen der Verlauf und die Prognose der Erkrankung sehr deutlich verbessert zu werden. Randomisierte Studiendaten zu dieser Therapie stehen nicht zur Verfügung und aufgrund der Seltenheit der Erkrankung erscheint eine solche Studie auch nicht durchführbar. Der Wirkungsmechanismus der intravenösen Immunglobulintherapie ist unklar. Vermutet werden immunmodulatorische und antiidiotypische Effekte der Behandlung. Die Therapie ist jedoch unspezifisch und teuer.
Das Wichtigste für die Praxis
• Das systemische «capillary leak»-Syndrom ist eine seltene und wenig bekannte Erkrankung mit rezidivierenden und potentiell lebensbedrohlichen Episoden einer kapillären Hyperpermeabilität, die zu einer Hypotonie, Hypalbuminämie, interstitiellen Ödemen und einer Hämatokriterhöhung durch Hämokonzentration führt.
• Typischerweise ist das Syndrom mit einer monoklonalen Gammopathie unklarer Signifikanz (MGUS) assoziert.
• Wegen der Seltenheit der Erkrankung und der unspezifischen Symptomen wird die Diagnose häufig erst lange Zeit nach Auftreten der ersten Symptome gestellt und ist möglicherweise unterdiagnostiziert.
• Therapeutisch müssen in der Akutphase die Hypotonie/Hypovolämie und die daraus resultierende Kreislaufinsuffizienz behoben werden.
• Zur Rezidivprophylaxe bietet sich eine Behandlung mit hochdosiertem intravenösen Immunglobulin an.
Disclosure statement
Die Autoren haben deklariert, keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag zu haben.
1 Clarkson B, Thompson D, Horwith M, Luckey EH. Cyclical edema and shock due to increased capillary permeability. Am J Med. 1960;29:193–216.
2 Kapoor P, Greipp PT, Schaefer EW, Mandrekar SJ, Kamal AH, Gonzalez-Paz NC, et al. Idiopathic Systemic Capillary Leak Syndrome (Clarkson’s Disease): The Mayo Clinic Experience. Mayo Clin Proc. 2010;85(10):905–12.
3 Pineton de Chambrun M, Gousseff M, Mauhin W, Lega JC, Lambert M, Rivière S, Dossier A, et al. Intravenous Immunoglobulins Improve Survival in Monoclonal Gammopathy-Associated Systemic Capillary-Leak Syndrome. Am J Med. 2017;130(10):1219.e19–1219.e27.
4 Druey KM, Greipp PR. Narrative Review: The Systemic Capillary Leak Syndrome. Ann Intern Med. 2010;153(2):90.
5 Doubek M, Brychtova Y, Tomiska M, Mayer J. Idiopathic systemic capillary leak syndrome misdiagnosed and treated as polycythemia vera. Acta hamatol. 2005; 113(2):150–1.