a Centre Universitaire de médecine générale et de santé publique (Unisanté), Lausanne; b Service de Dermatologie, Centre hospitalier universitaire vaudois (CHUV), Lausanne
Ein 50-jähriger Patient sucht die Notaufnahme auf, weil bei ihm vor zwei Monaten nicht juckende, purpurartige, runde, stellenweise tastbare, nicht konfluierende Hautläsionen an den unteren Extremitäten aufgetreten sind.
Fallbeschreibung
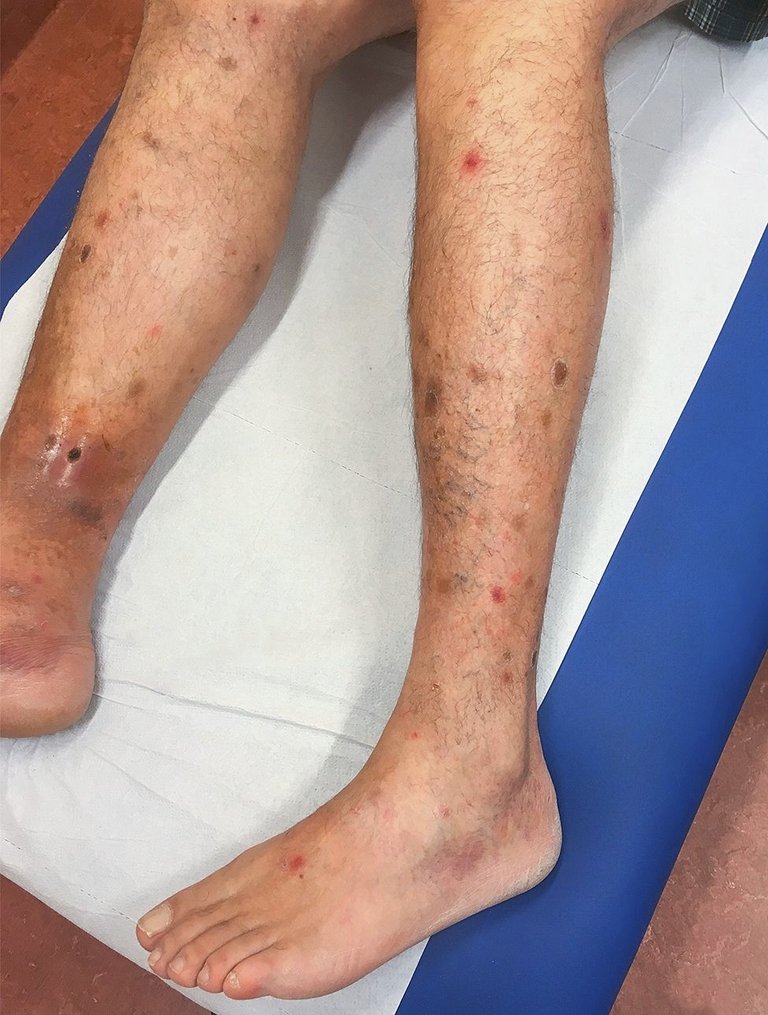
Ein 50-jähriger Patient in allgemein gutem Gesundheitszustand sucht die Notaufnahme auf, weil bei ihm vor zwei Monaten nicht juckende, purpurartige, runde, stellenweise tastbare, nicht konfluierende Hautläsionen an den unteren Extremitäten aufgetreten sind, wobei eine bei Palpation schmerzhafte nekrotische krustige Läsion am rechten Bein vorliegt (Abb. 1 sowie Abb. S1A im Online-Appendix dieses Artikels). Alte hyperpigmentierte Läsionen mit Anzeichen einer chronischen Veneninsuffizienz sind vorhanden. Der Rest der klinischen Untersuchung ist unauffällig.
Abbildung 1: Klinisches Bild: Purpurische Läsionen runder Morphologie mit einigen älter aussehenden hyperpigmentierten Läsionen und krustige Läsion am rechten Bein. Ein schriftlicher Informed Consent für die Publikation liegt vor.
Welche der Differentialdiagnosen, die bei diesem Krankheitsbild in Betracht gezogen werden müssen, ist zum jetzigen Zeitpunkt am wenigsten wahrscheinlich?
Eine gründliche Anamnese erlaubt es uns, einige dieser Diagnosen auszuschliessen. Der Patient erinnert sich nicht, durch Gliederfüsser gestochen worden zu sein, und klagt nicht über Juckreiz, überdies sind diese Läsionen im Allgemeinen einmalig und einseitig. Er erklärt, ungeschützten Geschlechtsverkehr ausschliesslich mit seiner Frau zu haben. Urogenitale Symptome liegen nicht vor. Die letzte Untersuchung auf sexuell übertragbare Infektionen ist jedoch schon mehrere Jahre her. Eine sekundäre Syphilis manifestiert sich ungefähr 3–10 Wochen nach der primären Inkubation und in Form eines stark variablen Hautausschlags. Eine leukozytoklastische Vaskulitis kann in Form palpabler purpurischer Läsionen, einige davon nekrotisch und überwiegend auf den unteren Extremitäten, vorliegen. Die PLEVA manifestiert sich mit polymorphen, oft makulopapulösen Läsionen mit einer feinen Desquamation und einer hämorrhagischen Nekrose. Eine lymphomatoide Papulose tritt in Form einer Eruption von Papeln von 0,5–1 cm Durchmesser, manchmal mehr, auf, die durch glimmerartige Schuppen bedeckt sind oder die hämorrhagisch und nekrotisch werden [1].
Welche zusätzliche Untersuchung ist am wenigsten relevant für die Differentialdiagnose?
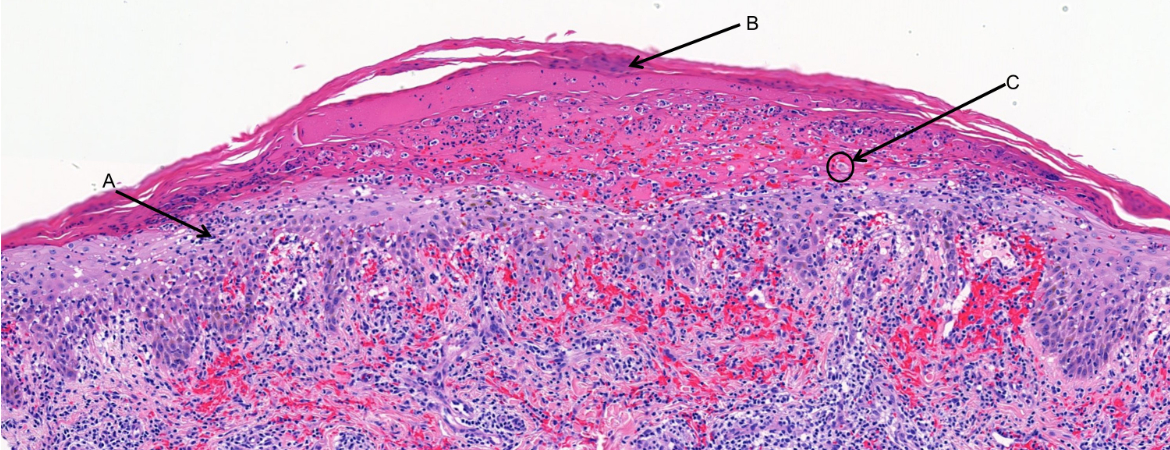
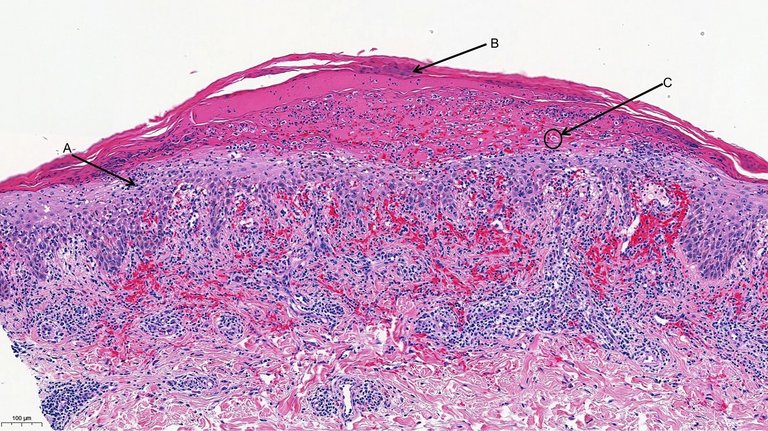
Eine Blutuntersuchung (vollständiges Blutbild, Nierenfunktion, Leberfunktion, Serologie für Hepatitis B und C, HIV und Syphilis, Elektrophorese der Serumproteine) bleibt unauffällig. Die ergänzenden Autoimmununtersuchungen ergaben einen normalen Wert für den Rheumafaktor, die Blutsenkungsgeschwindigkeit, antikernukleären Antikörper, ANCA-PR3, ANCA-MPO (Anti-Neutrophilen-Zytoplasma), Basalmembran-Antikörper, Kryoglobuline und Komplement-Antikörper C3 und C4. Das Urinsediment und der Albumin-Kreatinin-Quotient sind normal, was die Wahrscheinlichkeit einer Nierenerkrankung senkt. Eine Doppler-Sonographie der unteren Extremitäten zeigt beidseitig eine Insuffizienz der grossen Beinvenen und schliesst eine tiefe Venenthrombose aus. Wir hielten eine CT von Thorax und Abdomen in diesem Stadium nicht für sinnvoll, da diesuns in unserer Differentialdiagnose nicht weitergebracht hätte. Die Hautbiopsie an den Läsionen des linken Beins zeigte eine fokale Parakeratose, Dyskeratose, unregelmässige Akanthose mit Grenzflächenbefall und eine vakuoläre Degeneration der Basalschicht an mehreren Herden, isolierte Keratinozytennekrosen, ein entzündliches lymphohistiozytär-lichenoides Infiltrat der oberflächlichen Dermis und zahlreiche Herde mit extravasierten Thrombozyten (Abb. 2).
Abbildung 2: Histopathologischer Schnitt der ersten Hautbiopsie. 10-fache Vergrösserung, Färbung mit Hämatoxylin und Eosin. Die Markierung in der Abbildung beträgt 100 µm. A) Lichenoides Infiltrat der oberflächlichen Dermis. B) Parakeratose. C) Nekrotischer Keratinozyt.
Welche Diagnose stellen Sie?
Die Abwesenheit einer Schädigung der Gefässwand entkräftet die Diagnose Vaskulitis (ebenso wie ein negativer Autoimmunstatus). Das Fehlen eines dichten dermalen lymphozytären Infiltrats mit atypischen CD30+-Lymphozyten entkräftet auch die Diagnose einer lymphomatoiden Papulose. Anhand des Infektionsnachweises kann eine sekundäre Syphilis ausgeschlossen werden. Es gibt kein Argument für ein polymorphes Erythem, weder am klinischen Bild (keine kokardenförmigen Läsionen, keine palmoplantare Schädigung, keine Schädigung der Schleimhäute) noch an der Hautbiopsie. Das Resultat der Biopsie ist kompatibel mit der Diagnose einer PLEVA. Die lichenoide Pityriasis ist eine entzündliche dermatologische Erkrankung, die ein Spektrum an Erkrankungen von der akuten Form – der PLEVA – bis hin zur chronischen Form – der chronischen lichenoiden Pityriasis – umfasst. Die Ätiologie ist noch unklar. Die Erkrankung tritt am häufigsten zwischen dem 10. und 30. Lebensjahr auf und betrifft vor allem Männer mit einem Verhältnis von 1:2 [1]. Die Patientinnen und Patienten entwickeln rasch einen makulopapulösen Ausschlag mit einer feinen Desquamation, welcher sich zu purpurischen, hämorrhagischen und nekrotischen Papeln entwickeln kann. Die Läsionen sind oft polymorph und befinden sich in unterschiedlichen Entwicklungsstadien. Die betroffenen Körperteile sind im Allgemeinen der Rumpf und die unteren Extremitäten. Die Läsionen sind schmerzlos, können aber gelegentlich jucken und schmerzen. Sie können Folgeerscheinungen in Form von hypo- oder hyperpigmentierten Läsionen hinterlassen. Im Allgemeinen haben die Betroffenen keine anderen Symptome, können aber über Arthralgien, Bauchschmerzen, Fieber und Adenopathien berichten [1, 2].
Welche Behandlung ist am wenigsten geeignet?
Die Behandlung hängt vorrangig vom Schweregrad der Erkrankung ab. Die meisten Fälle weisen eine leichte Form der Erkrankung mit wenigen Symptomen auf, die zeitlich selbstlimitierend ist. In diesem Fall wird ein konservativer Zugang mit einer klinischen Überwachung und eventuell einer topischen Behandlung auf Grundlage von Kortikoiden vorgeschlagen. Bei einer Erkrankung mit ausgebreiteter und/oder persistierender (PLC) und/oder symptomatischer Schädigung wird eine systemische Behandlung vorgeschlagen. Die First-Line-Behandlung besteht in einer Antibiotikatherapie mit Tetrazyklin oder Erythromycin sowie einer Lichttherapie über 2–3 Monate. Ist diese nicht erfolgreich, kann eine Behandlung mit Methotrexat verschrieben werden. Es ist jedoch wichtig zu erwähnen, dass es bis heute nur wenige Studien zu systemischen Behandlungen gibt, was auf die Seltenheit der Erkrankung und die unterschiedliche Dauer bis zur Spontanheilung zurückzuführen ist [2, 3].
Unser Patient wurde zur Symptomlinderung vier Wochen lang mit topischen Kortikoiden behandelt.
Welche der folgenden Aussagen ist in Bezug auf die Prognose einer PLEVA korrekt?
Der Krankheitsverlauf ist unterschiedlich und dauert von einigen Wochen über Monate bis hin zu Jahren mit einem hohen Rezidivrisiko [1, 4]. Die Läsionen tendieren dazu, zeitlich selbstlimitierend zu sein, und eine Spontanheilung ist häufig. Es gibt jedoch eine seltene Form – die «febrile ulzeronekrotische Mucha-Habermann-Krankheit» – bei der die schnelle Bildung von ulzero-bullösen Läsionen auftritt, die sich zusätzlich infizieren können und mit anderen Organenschäden verbunden ist, was eine hohe Mortalitätsrate zur Folge hat.
Eine der Hypothesen zur Ätiologie der Pityriasis Lichenoides ist, dass sie Teil einer proliferativen Störung klonaler T-Zellen ist und die Immunantwort des Wirts entscheidend für die Progression zu einem kutanen Lymphom ist. Diese Hypothese bleibt bislang jedoch umstritten [1, 2].
Unser Patient wird sieben Monate später erneut gesehen und zeigt eine günstige Entwicklung. Während der Nachsorge stellen wir postinflammatorisch aussehende hyperpigmentierte makuläre Läsionen ohne aktive Läsionen fest (Abb. S1B und S1C im Online-Appendix). Somit bestand keine Notwendigkeit einer systemischen Behandlung.
Diskussion
Die PLEVA ist eine seltene Erkrankung, deren Ätiologie noch unklar ist. An diese Diagnose sollte bei jeder polymorphen, schuppigen, makulo-papulären Hautschädigung mit geringen Symptomen gedacht werden. Die wichtigsten Differentialdiagnosen sind: lymphomatoide Papulose, leukozytoklastische Vaskulitis, polymorphes Erythem, sekundäre Syphilis, Tropfpsoriasis und das Gianotti-Crosti-Syndrom (Tab. 1).
Tabelle 1: Differentialdiagnosen einer PLEVA, zu berücksichtigen bei jeder Person mit einer polymorphen Hautschädigung makulopapulöser squamöser Art [1, 5]
Häufigkeit
Merkmale
Inflammatorisch
Polymorphes Erythem
Häufig
Kokardenförmige Läsionen, oft an den Gliedmassen Schädigung der Schleimhäute möglich Reaktion auf einen infektiösen Erreger (häufig Herpes-simplex-Virus und Mycoplasma pneumoniae)
Lichenoides Arzneimittelexanthem
Selten
Kenntnis der eingenommenen Arzneimittel
Leukozytoklastische Vaskulitis
Selten
Purpura vorwiegend an den unteren Gliedmassen Gelegentlich systemische Symptome
Tropfpsoriasis
Etwa 0,2%
Generalisierte multiple Läsionen, klein, ovalär und squamös Pruritus Oft nach einer Infektion mit Streptococcus beta-haemolyticus
Gianotti-Crosti-Syndrom
Selten, betrifft vor allem Kinder
Diffuses Exanthem Assoziiert mit einer Infektion mit dem Epstein-Barr-Virus
Dermatitis herpetiformis
10–75/100 000
Symmetrischer papulovesikulärer Ausschlag auf der Oberfläche der Extensoren Pruritus Häufig assoziiert mit Zöliakie
Tumoral
Lymphomatoide Papulose
Selten
Generalisierte papulonoduläre Läsionen
Infektiös
Sekundäre Syphilis
6,3 auf 100000
Makulopapulöse Läsion, palmoplantare Schädigung häufig Systemische Symptome Ungeschützter Geschlechtsverkehr
Rickettsiose
Endemische Gebiete
Reise mit bekanntem Insektenstich Rash mit zentripetalem Ausschlag Systemische Symptome Palmoplantar keine Symptome
Hauttuberkulose
Selten, vor allem in tropischen Ländern
Die Klinik hängt von der Infektionsstelle ab Infektion parallel mit einer Lungentuberkulose
Varicella
Häufig, vor allem bei Kindern
Rasches Auftreten eines papulovesikulären Ausschlags in verschiedenen Entwicklungsstadien Pruritus, erhöhte Kontagiosität Systemische Symptome
PLEVA: Pityriasis lichenoides et varioliformis acuta.
Die Diagnose wird durch eine Kombination aus dem klinischen Bild (auch dem klinischen Verlauf) und den in der Hautbiopsie gefundenen Elementen gestellt, die die Diagnose lenken können. Die Behandlung ist je nach Schwere der Erkrankung unterschiedlich (Tab. S1 im Online-Appendix dieses Artikels). Bei einem leichten Befall wird eine konservative Behandlung vorgeschlagen. Die Dauer des Verlaufs kann vonWochen über Monate bis hin zu Jahren variieren und die Läsionen tendieren dazu, zeitlich selbstlimitierend zu sein [1].
Frage 1: a. Frage 2: e. Frage 3: a. Frage 4: a. Frage 5: e.
Dr. med. Aline Thorens
Centre Universitaire de médecine générale et de santé publique (Unisanté), Lausanne
Verdankung
Die Autoren möchten Prof. Daniel Hohl für die Aufsicht des Befunds der dermatopathologischen Untersuchung danken.
Korrespondenz
Dr. med. Aline Thorens
Centre Universitaire de médecine générale et de santé publique (Unisanté)
Rue du Bugnon 44
CH-1011 Lausanne
Literatur
1 Fernandes NF, Rozdeba PJ, Schwartz RA, Kihiczak G, Lambert WC. Pityriasis lichenoides et varioliformis acuta: a disease spectrum. Int J Dermatol. 2010;49(3):257–61.
2 Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55(4):557–72.
3 Bellinato F, Maurelli M, Gisondi P, Girolomoni G. A systematic review of treatments for pityriasis lichenoides. J Eur Acad Dermatol Venereol. 2019;33(11):2039–49.
4 Ediale C, Felix K, Anderson K, Ahn C, McMichael AJ. An Atypical Presentation of PLEVA: Case Report and Review of the Literature. J Drugs Dermatol JDD. 2019;18(7):690–1.
5 Wolff K, Johnson RA, Suurmond D. Fitzpatrick atlas en couleurs de dermatologie clinique. Paris: Médecine – sciences Flammarion; 2005.