Eine 57-jährige Patientin wurde mit ausgedehnter Livedo racemosa an den unteren Extremitäten und im Wangenbereich vorstellig. B-Symptomatik sowie artikuläre oder neurologische Symptomatik fehlten.
Hintergrund
Der Begriff «Livedo» beschreibt medizinisch eine erythematös-livide Verfärbung der Haut, die durch eine fokale Reduktion oder Unterbrechung des Blutflusses in den dermalen Arterien und Arteriolen entsteht. In der klinischen Untersuchung ist die Livedo reticularis mit lividen, geschlossenen, netzartigen Hautzeichnungen von der Livedo racemosa mit ebenfalls lividen, jedoch diskontinuierlichen und Blitzfigur-ähnlichen Hautzeichnungen abzugrenzen. Die Livedo reticularis tritt häufig ohne Krankheitswert vor allem bei jungen Frauen als Cutis marmorata auf, gelegentlich aber auch im Rahmen einer Systemerkrankung, zum Beispiel bei Konnektivitiden. Sie betrifft mehrheitlich die unteren Extremitäten und ist stress- und/oder temperaturabhängig [1]. Im Gegensatz zur Livedo reticularis ist die Livedo racemosa immer Ausdruck einer strukturellen Pathologie. Die Veränderungen an der Haut können dabei den übrigen Manifestationen einer Grunderkrankung Monate bis Jahre vorausgehen. Die häufigsten mit Livedo racemosa assoziierten Systemerkrankungen sind ein primäres oder sekundäres Antiphospholipid-Syndrom, letzteres bei systemischem Lupus erythematodes. Seltenere Ursachen für eine Livedo racemosa sind zum Beispiel die Livedovaskulopathie und das Sneddon-Syndrom (Symptomtrias: Kopfschmerzen, Livedo racemosa und rezidivierende zerebrovaskuläre Ischämien/Infarkte) [1, 2].
Fallbericht
Anamnese
Wir berichten über eine 57-jährige Patientin mit ausgedehnter Livedo racemosa an den unteren Extremitäten und im Wangenbereich. B-Symptomatik sowie artikuläre oder neurologische Symptomatik fehlten. Ein vorangehender viraler Infekt der oberen Atemwege hatte zur Einnahme von Acetylsalicylsäure geführt. In der persönlichen Anamnese fielen drei vorangegangene Episoden einer Livedo reticularis an den unteren Extremitäten mit jeweils spontaner Remission auf. Als potentielle Auslöser dieser Vorepisoden kamen retrospektiv virale Infekte und die Exposition gegenüber östrogenhaltigen Vaginalsuppositorien infrage. In der Anamnese waren zudem zwei Spontanaborte in der 12. beziehungsweise 14. Schwangerschaftswoche eruierbar. In der Familienanamnese fand sich ein schwerer, ätiologisch ungeklärter ischämischer Insult beim 42-jährigen Bruder.
Status und Befunde
In der klinischen Untersuchung imponierten die oben erwähnten kutanen Befunde (Abb. 1), ohne Nekrosen und ohne Schleimhautbeteiligung.
Abbildung 1: Livedo racemosa im Gesässbereich (A ) und am Oberschenkel beidseits (B ).
Die restliche internistische und neurologische Untersuchung ergab keine pathologischen Befunde. Die Vitalwerte waren normal (Temperatur 37,3 °C, Blutdruck 140/85 mm Hg, Puls 75/min, Sauerstoffsättigung [SpO2] unter Raumluft 98%).
Das Blutbild zeigte sich unauffällig, ebenso Gerinnungsstatus, Kreatinin und C-reaktives Protein (CRP). Die erweiterten Laboruntersuchungen ergaben positive Antiphospholipid-Antikörper (aPL) vom Typ Anti-Cardiolipin-(aCL-)IgM (33 IgM-Antiphospholipid-Einheiten [MPL-U]/ml, Norm <10 MPL-U/ml) und Anti-β2-Glykoprotein-I-(anti-β2GPI-)IgM (18 U/ml, Norm <7 U/ml), wohingegen die Antikörper Anti-aCL-IgG (0,6 GPL-U/ml, Norm <10 GPL-U/ml), Anti-β2GPI-IgG (0,7 U/ml, Norm <7 U/ml) und Lupus-Antikoagulans (LA) negativ waren. Die Vaskulitisserologien (Antinukleäre Antikörper [ANA], Anti-Neutrophile cytoplasmatische Antikörper [ANCA] mit Proteinase-3-[PR3-] und Myeloperoxidase-[MPO-]Spezifität, Doppelstrang-Desoxyribonukleinsäure-[ds-DNA-]Antikörper, Sjögren-Syndrom-Antigen-Typ-A[SSA/Ro]- und -B[SSB/La]-Antikörper), Rheumafaktoren und Komplementfaktoren (C3, C4) waren unauffällig. Kryoglobuline, Serumelektrophorese/-Immunfixation und Hepatitis-B- und -C-Serologien waren nicht nachweisbar respektive normalwertig.
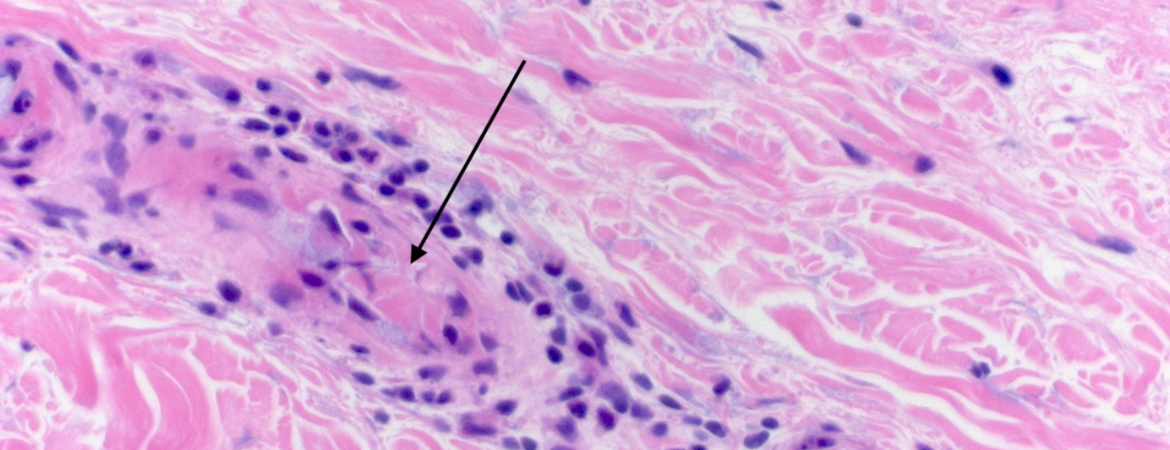
Die Hautbiopsie im Livedobereich des ventralen Oberschenkels zeigte eine mikrothrombotische Angiopathie mit Verschlüssen zahlreicher dermaler Kapillaren und geringer lymphozytärer und granulozytärer Entzündungsreaktion ohne Hämorrhagien oder Beteiligung von Arterien oder Venen (Abb. 2).
Abbildung 2: Hautbiopsie; Hämatoxylin-Eosin-Färbung. Gefässbezogene Zellinfiltrate in der Dermis (A , 50-fache Vergrösserung); Mikrothromben in den Kapillaren (Pfeil in B , 400-fache Vergrösserung). Abbildungen freundlicherweise zur Verfügung gestellt von der Pathologie des Luzerner Kantonsspitals.
Diagnose
Zusammenfassend bestand damit der Verdacht auf ein Antiphospholipid-Syndrom (APS): die klinischen Kriterien für ein «obstetric APS» (zwei Spontanaborte in der Frühschwangerschaft) und für ein «non-criteria thrombotic APS» (Livedo racemosa) waren erfüllt, das Laborkriterium mit der benötigten Folgebestimmung nach frühestens drei Monaten jedoch noch nicht. Die Kontrolle der aCL-IgM-Antikörper (43 MPL-U/ml) und anti-β2GPI-IgM-Antikörper (21 U/ml) drei Monate nach der Erstuntersuchung fielen im gleichen Titerbereich positiv aus, entsprechend einem niedrigen bis moderaten Antikörper-Risikoprofil. Mit diesem Befund war auch das Laborkriterium gemäss den überarbeiteten Sapporo-Klassifikationskriterien (Tab. 1) erfüllt, sodass – gemeinsam mit dem klinischen und histologischen Befund – die Diagnose eines APS endgültig gestellt werden konnte.
Eine oder mehrere Episoden von arteriellen, venösen oder mikrovaskulären Thrombosen in beliebigen Organen und Geweben. Bestätigung durch bildgebende oder histopathologische Verfahren. Kein Nachweis einer Vaskulitis in der Histopathologie.
Schwangerschaftsmorbidität
a) Eine oder mehrere Totgeburten in oder nach der 10. Schwangerschaftswoche (SSW) eines morphologisch normalen Fetus. b) Eine oder mehrere Frühgeburten eines morphologisch normalen Neugeborenen vor der 34. SSW, aufgrund von Eklampsie, schwere Präeklampsie oder Plazentainsuffizienz. c) Drei oder mehr Spontanaborte vor der 10. SSW bei Ausschluss anatomischer, hormoneller und chromosomaler Ursache.
Laborkriterien
1. Lupus Antikoagulans
Nachweis von Lupus Antikoagulans (LA) im Plasma durch eine verlängerte phospholipid-abhängige Gerinnungszeit unter Verwendung verschiedener Testprinzipien. und/oder
2. Anti-Cardiolipin-Antikörper
Erhöhte Anti-Cardiolipin-Antikörper (aCL) des IgG- und/oder IgM-Isotyps im Serum oder Plasma mit mittlerem oder hohem Titer (≥40 U/ml [GPL oder MPL] oder Titer ≥99. Perzentile), gemessen mit ELISA-Test. und/oder
3. Anti-β2-Glykoprotein-I-Antikörper
Erhöhte Anti-β-2-Glykoprotein-Antikörper (anti-β2GPI) des IgG- und/oder IgM-Isotyps im Serum oder Plasma (Titer ≥99. Perzentile), gemessen mit einem ELISA-Test.
4. Zeitraum
Nachweis an zwei oder mehr Zeitpunkten im Abstand von mindestens 12 Wochen.
Eine therapeutische Antikoagulation mit dem Ziel der Risikoreduktion einer arteriellen oder venösen Thrombose wurde mit niedermolekularem Heparin 5000 IE subkutan zweimal täglich (bei einem Körpergewicht von 63 kg) für insgesamt drei Wochen eingeleitet und anschliessend überlappend auf Acetylsalicylsäure 100 mg peroral einmal täglich umgestellt. Zeitgleich erfolgte eine lokale Kompressionstherapie (Kompressionstrümpfe der Klasse 2) der unteren Extremitäten über 24 Stunden.
Bei initial deutlich entzündlicher Reaktion im Bereich einzelner Racemosa-Läsionen mit Nekrosegefahr, noch ausstehendem Hautbiopsiebefund und differentialdiagnostischem Verdacht auf eine Vaskulitis erfolgte die einmal tägliche Gabe von Prednisolon 40 mg peroral für drei Tage mit raschem Ausschleichen nach Erhalt des Biopsiebefundes. Unter der genannten Therapie kam es zu einer schnellen Besserung des Hautbefundes (Abb. 3). In der Verlaufskontrolle nach drei Monaten waren sämtliche Läsionen vollständig und ohne Pigmentverschiebungen oder Atrophien abgeheilt.
Diskussion
Hautbiopsien bei Livedo racemosa zeigen histopathologisch in kleinen und/oder mittelgrossen Blutgefässen der Dermis und Subcutis entweder intraluminale Thromben, eine subintimale Gefässwandverdickung oder eine Vaskulitis. Im vorliegenden Fall stand eine mikrothrombotische Angiopathie im Vordergrund. Normalbefunde durch «sampling error» sind durch eine optimale Biopsielokalisation, eine ausreichende Biopsiegrösse und durch Mehrfachbiopsien vermeidbar. Die Livedo racemosa ist die häufigste Hautmanifestation bei Menschen mit APS und wurde in bis zu 40% der Fälle als Erstsymptom beobachtet – bei an APS Erkrankten mit systemischem Lupus erythematodes (SLE) sogar in bis zu 70% der Fälle [2].
Beim APS führen die Antiphospholipid-Antikörper, eine heterogene Gruppe von Autoantikörpern gegen Phospholipide und an Phospholipide gebundenen Proteinen, zu einer erworbenen Thrombophilie. Die Pathogenese dieser Thrombophilie ist bisher nicht vollständig geklärt. Einerseits werden Triggerfaktoren wie Infektionen, östrogenhaltige Pillen, Nikotin und Operationen postuliert, die oxidativen Stress und Endothelschäden induzieren, andererseits scheint die Aktivierung von Komplement und Endothelzellen durch die oxidative Formänderung des β2GPI mit Freilegung immunogener Epitope (Domäne I [DI]) zur Andockung der DI-spezifischen anti-β2GPI-Antikörper eine wichtige Rolle zu spielen. Andere Pathomechanismen, auf die hier nicht eingegangen wird, führen zu den Schwangerschaftskomplikationen beim APS [3, 4].
Die Lebensprävalenz des APS in der Bevölkerung liegt bei 1–5%, Frauen sind häufiger betroffen. Das APS kann primär (ohne Grunderkrankung) oder sekundär in Assoziation mit anderen Autoimmunerkrankungen, insbesondere dem SLE, sowie mit Infektionen, Medikamenten oder Neoplasien auftreten [3].
Klinische Hauptmanifestationen sind wiederkehrende mikrovaskuläre, venöse und arterielle Thromben in verschiedensten Organen und Geweben («thrombotic APS») sowie Schwangerschaftskomplikationen («obstetric APS»). Eine Zusammenfassung ergibt sich aus den überarbeiteten Sapporo-Klassifikationskriterien (Tab. 1). Weitere klinische Manifestationen des APS sind beispielsweise Livedo reticularis/racemosa, Thrombozytopenie, APS-Nephropathie oder -Valvulopathie («non-criteria thrombotic APS»). APS-definierende Laborkriterien sind der Nachweis von aPL als LA und/oder als aCL- und anti-β2GPI-Antikörper zu zwei Zeitpunkten im Abstand von mindestens zwölf Wochen (Tab. 1) [3, 5].
Mit dem vorliegenden Fall wollen wir die Wichtigkeit der Gesamtbeurteilung von klinischen Kriterien, hier am Beispiel der Livedo racemosa (als seltene Manifestation und nicht thrombotisches Kriterium) und der schwangerschaftsassoziierten Komplikationen, und Laborkriterien zur Diagnosestellung des APS darstellen. Die Histologie krönt den Fall auf pathohistologischer Ebene – von uns vor allem durch die Abbildungen dargestellt.
In der Literatur gibt es unterschiedliche Angaben, ab welchem Wert das Resultat eines Immunassays, insbesondere des aCL-Antikörper-Tests, als positiv zu werten ist. Diese Unsicherheit ist Folge verschiedener immunologischer Bestimmungsmethoden und führte dazu, dass der Cut-off für einen positiven Befund bei vielen Tests auf 40 U/ml gesetzt wurde. Seit der Einführung und Weiterverbreitung von automatisierten Testsystemen und nach Durchführung von Multizenter-Laborstudien hat sich klar gezeigt, dass Werte über der 99. Perzentile – oder sogar über der 95. Perzentile – eines Normalkollektivs als positiv zu werten sind. Werte über 40 oder 50 U/ml gelten als stark positiv [6]. Mit den aktuellen Assays sind somit aCL-Antikörper mit einem Wert von ≥10 U/ml (GPL oder MPL) und anti-β2GPI-Antikörper ≥7 U/ml als positiv zu werten. Zu betonten ist, dass positive aPL-Antikörper-Werte ohne klinische Manifestation nicht zur Diagnose eines APS genügen, da das klinische Kriterium fehlt.
Die Wertigkeiten der einzelnen aPL-Antikörper stehen aufgrund der heterogenen Analytik und des Studienkollektivs seit Jahren zur Diskussion. Eine fallbezogene Interpretation der einzelnen aPL-Antikörper ist zu empfehlen.
Entsprechend den konkreten Resultaten der diversen aPL-Antikörper-Tests wird ein Antikörper-Risikoprofil mit niedrigem, moderatem und hohem Risiko definiert (Tab. 2) [7].
Anhand des aPL-Risikoprofils und der klinischen Manifestationen («thrombotic», «non-criteria thrombotic» und/oder «obstetric» APS, asymptomatisch) erfolgt die Wahl der entsprechenden Therapie. Vereinfacht zusammengefasst ist die orale Antikoagulation mit Vitamin-K-Antagonisten (VKA) nach wie vor die Standardtherapie beim «thrombotic APS» (venöse oder arterielle Thrombosen) mit einer Ziel-International-Normalized-Ratio (INR) von 2 bis 3. Der Einsatz direkter oraler Antikoagulanzien (DOAK) als Alternative zu VKA kann bei venösen Thrombosen und niedrigem aPL-Risikoprofil erwogen werden. Die Antikoagulation sollte fortgeführt werden, solange die aPL nachweisbar sind (Kontrolle alle zwölf Monate empfohlen). Ein Stopp der Antikoagulation kann erwogen werden, wenn die aPL zweimal im Abstand von mindestens sechs Monaten nicht mehr nachweisbar sind [7]. In den Behandlungsempfehlungen der «European Alliance of Associations for Rheumatology» (EULAR) wird bei unprovozierter Thromboembolie bisher weiterhin eine Dauerantikoagulation empfohlen [8]. Bei asymptomatischen Menschen mit hohem oder moderatem aPL-Risikoprofil und bei Frauen mit durchgemachtem «obstetric APS» wird ausserhalb der Schwangerschaft eine Primärprophylaxe mit Acetylsalicylsäure 100 mg empfohlen. Zudem ist in allen asymptomatischen Fällen eine venöse Thromboseprophylaxe in Risikosituationen (wie Immobilisation, Operationen) empfohlen [7]. Begleitend gilt für alle Risikokategorien eine strikte Kontrolle der kardiovaskulären Risikofaktoren, die Elimination potentieller Triggerfaktoren (Nikotin, Östrogensubstitution) und die Therapie einer assoziierten Grunderkrankung (wie SLE) [3, 4].
Das Wichtigste für die Praxis
Die Livedo racemosa ist immer Ausdruck einer organischen Gefässproblematik und bedarf einer Notfallabklärung.
Die Diagnose Livedo racemosa erfolgt klinisch (typischer Hautbefund) und histologisch.
Das Antiphospholipid-Syndrom kann multiple klinische Manifestationen mit sich bringen: thrombotisch, nicht thrombotisch (hier am Beispiel der Livedo racemosa), schwangerschaftsassoziiert.
Der zur Diagnose eines Antiphospholipid-Syndroms zusätzlich notwendige Nachweis von Antiphospholipid-Antikörpern (Anti-Cardiolipin-, Anti-β-2-Glykoprotein-I-Antikörper und/oder Lupus-Antikoagulans) muss zweimal in einem Abstand von mindestens zwölf Wochen positiv sein.
Die Therapie des Antiphospholipid-Syndroms richtet sich nach klinischer Manifestation, Antiphospholipid-Antikörper-Risikoprofil und assoziierten Autoimmunerkrankungen.
Dr. med. univ. Andrea Röthlin
Klinik für Innere Medizin, Luzerner Kantonsspital, Luzern
Verdankung
Wir danken Prof. Dr. Dr. med. Walter Alfred Wuillemin, Senior Consultant der Hämatologie, Dr. med. Lukas Schmid, Chefarzt der Rheumatologie, und Prof. Dr. med. Joachim Diebold, Chefarzt der Pathologie, des Kantonsspitals Luzern für die aufmerksame Durchsicht des Artikels und ihre Anregungen.
Informed Consent
Ein schriftlicher Informed Consent zur Publikation liegt vor.
Korrespondenz
Dr. med. univ. Andrea Röthlin
Ärzte Hinwil
Walderstrasse 10
CH-8340 Hinwil
Literatur
1 Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol. 2006;33(12):2379–82.
2 Pincelli MS, Echavarria AMJ, Criado PR, Marques GF, Morita TCAB, Valente NYS, de Carvalho JF. Livedo racemosa: Clinical, laboratory, and histopathological findings in 33 patients. Int J Low Extrem Wounds. 2021;20(1):22–8.
3 Linnemann B. Antiphospholipid syndrome – an update. Vasa. 2018;47(6):451–64.
4 Arachchillage D, Laffan M. Pathogenesis and management of antiphospholipid syndrome. Br J Haematol. 2017;178(2):181–95.
5 Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid-syndrome (APS). J Thromb Haemost. 2006;4(2):295–306.
6 Devreese KMJ, Ortel TL, Pengo V, de Laat B; Subcommittee on Lupus Anticoagulant/Antiphospholipid antibodies. Laboratory criteria for antiphospholipid syndrome: communication from the SSC of the ISTH. J Thromb Haemost. 2018;16(4):809–13.
7 Kalmanti L, Lindhoff-Last E. Treatment of vascular thrombosis in antiphospholipid syndrome: An update. Hamostaseologie. 2020;40(1):31–7.
8 Tektonidou MG, Andreoli L, Limper M, Amoura Z, Cervera R, Costedoat-Chalumeau N, et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheum Dis. 2019;78(10):1296–1304.