

Publié le 14.06.2017
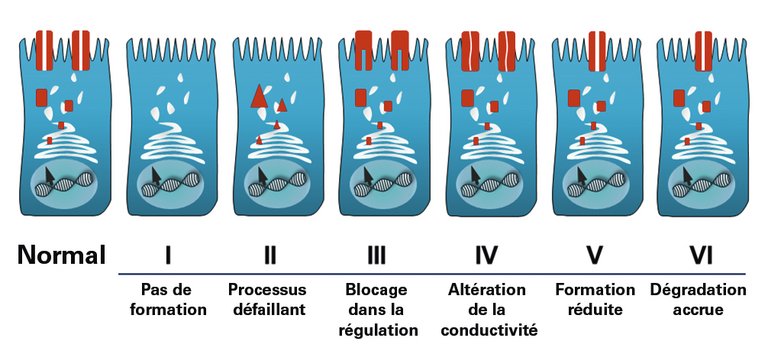
| Tableau 1:Symptômes cliniques et complications de la mucoviscidose (MV) en fonction de l’âge. Les pourcentages entre parenthèses donnent la prévalence en Suisse en 2014 pour autant que les données soient disponibles (d’après le rapport annuel 2014 ECFSPR). | ||
| Age | Présentation clinique | |
| Respiratoire | Gastro-intestinale et autres | |
| Indépendamment de l‘âge | – Toux productive – Production d’expectorations – Infections des voies respiratoires avec pathogènes typiques (Staphyloccocus aureus, Haemophilus influenzae, Pseudomonas aeruginosa) | – Stagnation du poids – Stéatorrhée – Diarrhée – Ballonnements – Douleurs abdominales – Carence en vitamines liposolubles – Alcalose métabolique hypochlorémique (syndrome pseudo-Bartter, syndrome de perte de sel) |
| Nouveau-nés | – Iléus méconial (15,6%) – Atrésie intestinale – Hyperbilirubinémie prolongée | |
| Enfants | – Toux récidivante / chronique – Pneumonies | – Prolapsus rectal – Syndrome d’obstruction intestinale distale (SOID) – Invagination – Cholestase – Hypoprotéinémie (également avec œdèmes) – Déshydratation – Anémie |
| Adolescents & adultes | Comme les enfants, plus: – Bronchiectasies – Hémoptysies – Pneumothorax – Aspergillose bronchopulmonaire allergique (ABPA, 5,7%) – Infection par mycobactéries atypiques – Hippocratisme digital, ongles en verre de montre – Rhinosinusite chronique – Polypose naso-sinusienne – Anosmie | Comme les enfants, plus: – Pancréatite aiguë – Hépatopathie (23,0%) – Hypertension portale – Diabète sucré associé à MV (DAMV; 23,7% pour les ≥18 ans) – Ostéoporose – Arthrite associée à la MV – Absence congénitale bilatérale des canaux déférents (CBAVD) |
| ECFSPR = «European Cystic Fibrosis Society Patient Registry» | ||
| Tableau 2:Valeurs de référence pour les différentes méthodes de test de la sueur. | ||
| Concentration de chlorure | Conductivité (équivalents NaCl) | |
| MV | ≥60 mmol/l | ≥80 mmol/l |
| Zone grise | 30–59 mmol/l | 50–79 mmol/l |
| Pas de MV | <30 mmol/l | <50 mmol/l |
| Volume de sueur | ≥15 µl (Macroduct®) | ≥3 µl (Nanoduct®) |
| Taux de sueur | ./. | ≥1,0 g/m2/min |
| MV = mucoviscidose, ./. = non applicable | ||

| Tableau 3: Valeurs de référence cliniques de la mucoviscidose en fonction des groupes d’âge en Suisse en 2014 (d’après le rapport annuel 2014 ECFSPR). Les données microbiologiques portent sur la prévalence d’une infection pulmonaire chronique. | ||
| <18 ans | ≥18 ans | |
| VEMS vs. valeurs prédites1,2 | 89,2% (77,4, 100,5) | 62,1% (46,9, 81,8) |
| Poids (z-scores) | –0,4 (–1,1, 0,3) | –0,5 (–1,2, 0,1) |
| P. aeruginosa | 12,8% | 53,1% |
| S. aureus | 55,2% | 49,2% |
| MNT3 | 0,8% | 6,5% |
| 1 «Global Lung Initiative equations»; tous les patients à partir de 6 ans sans transplantation pulmonaire 2 Médiane (25e, 75e percentiles) 3 MNT = mycobactéries non tuberculeuses ECFSPR = «European Cystic Fibrosis Society Patient Registry» | ||
| Tableau 4: Programme de surveillance standardisé en centre de MV (modifié selon [5]); ici à l’exemple de l’hôpital pour enfant de Zurich. | ||
| Examen | Détails | Fréquence |
| Instruction de physiothérapie | Y compris vérification de la technique d’inhalation; en centre ou dans un cabinet spécialisé | Hebdomadaire à bimensuelle |
| Anamnèse | Médecin spécialiste de la MV | Tous les 3 mois |
| Contrôle thérapeutique | Médecin spécialiste de la MV; y compris information sur la supplémentation en sel | |
| Examen physique | Médecin spécialiste de la MV | |
| Gestion de la maladie | Infirmière spécialiste de la MV | |
| Formation des patients | Y compris hygiène; médecin spécialiste de la MV et infirmière spécialiste de la MV | |
| Etat nutritionnel | Poids, taille, indice de masse corporelle, poids-pour-taille | |
| Fonction pulmonaire | Spirométrie, pléthysmographie corporelle; à partir de 4 ans | |
| «Multiple-Breath Washout» | Indice de clairance pulmonaire (LCI); à partir de 3 ans | |
| Microbiologie | Expectorations, ou frottis pharyngé | |
| Conseil social | Assurances, finances, droit | |
| Prélèvements sanguins annuels | Hémogramme, électrolytes, oligoéléments, fer, ferritine, vitamines liposolubles A/D, protéine totale, albumine, enzymes hépatiques, statut de la coagulation, paramètre de cholestase, acide biliaire, glycémie à jeun, HbA1c, paramètres rénaux, ImmunoCAP (s×1, m×1) | Annuelle |
| Hyperglycémie provoquée per os | A partir de 10 ans | |
| Echographie abdominale | ||
| Conseil nutritionnel | Y compris contrôle de la supplémentation enzymatique | |
| Bronchoscopie avec LBA | Chez les patients ne produisant pas d’expectorations ou si nécessaire | |
| Dépistage MNT | A partir du LBA ou des expectorations; à partir de 10 ans | |
| Avis ORL | Avec endoscopie nasale | |
| Ostéodensitométrie | A partir de 12 ans | Tous les 2 ans |
| Echocardiographie | A partir de 16 ans | |
| IRM pulmonaire | De façon routinière ou au besoin | Tous les 4 ans |
| Sérologie ABPA | IgE totales, IgE spécifiques, antigènes recombinants | Au besoin |
| Radiographie thoracique | ||
| Tomodensitométrie thoracique | ||
| Audiométrie | En cas d’administration i.v. répétée d’aminosides | |
| Conseil psychologique | ||
| MV = mucoviscidose, LBA = lavage broncho-alvéolaire, MNT = mycobactéries non tuberculeuses, ABPA = aspergillose broncho-pulmonaire allergique | ||
| Tableau 5: Stratégies thérapeutiques pour le traitement de la mucoviscidose (MV) incluant les données disponibles relatives à la fréquence en Suisse en 2014 (d’après le rapport annuel 2014 ECFSPR). | |||
| Stratégie | Formes thérapeutiques | Principaux objectifs thérapeutiques | Fréquence en suisse1 |
| Traitement nutritionnel | Enzymes pancréatiques | Développement normal du poids et de la taille | 88,9% |
| Alimentation hautement calorique | ./.2 | ||
| Vitamines liposolubles | Prévention de l’hypovitaminose, carence en vitamine K et ostéoporose | ./. | |
| Supplémentation orale en chlorure de sodium | Prévention d’un syndrome de perte de sel | ./. | |
| Traitement des complications gastro-intestinales | Acide ursodésoxycholique | Traitement de la cholestase; prévention d’une hépathopathie | 29,8% |
| Insuline | Traitement du DAMV | 23,7%3 | |
| Macrogol | Traitement et prévention du SOID | ./. | |
| Sécrétolyse | Physiothérapie respiratoire (avant tout drainage autogène et aide à la sécrétolyse) | Amélioration de la clairance mucociliaire; prévention des atélectasies, bronchiectasies et infections pulmonaires / exacerbations | ./. |
| Inhalation d’une solution hypertonique chlorure de sodium | 60,9% | ||
| Inhalation de rhDNAse | 38,2% | ||
| Bronchodilatation | Bêta-2-mimétiques (à courte et longue durée d’action) | Traitement préliminaire avant solution hypertonique
chlorure de sodium; traitement du trouble respiratoire obstructif chronique | 88,7% |
| Traitement de l’infection pulmonaire | Antibiotiques systémiques | Traitement de l’exacerbation pulmonaire; prévention d’une perte de la fonction pulmonaire | |
| Antibiotiques inhalés | Eradication de P. aeruginosa; suppression en cas d’infection chronique à pseudomonas pour la prévention des exacerbations | 34,7% | |
| Macrolides (avant tout azithromycine en cas d’infection chronique à pseudomonas) | Amélioration de la fonction pulmonaire (effet anti-inflammatoire supposé) | 28,3% | |
| Antimycosiques systémiques | Traitement de l’ABPA | ./. | |
| Corticoïdes systémiques | ./. | ||
| Clairance des sécrétions naso-sinusiennes | Rinçage nasal, inhalation sinusienne | Prévention / traitement de la polypose naso-sinusienne | ./. |
| Correction du défaut de base | Correcteurs et potentiateurs de CFTR | Amélioration / stabilisation de la pneumopathie; prise de poids | <1% |
| 1 en % de tous les patients MV 2 non documenté 3 chez les patients ≥18 ans MV = mucoviscidose, ECFSPR = «European Cystic Fibrosis Society Patient Registry», DAMV = diabète associé à la fibrose kystique, SOID = obstruction intestinale distale, ABPA = aspergillose broncho-pulmonaire allergique | |||
Publié sous la licence du droit d'auteur.
"Attribution - Non-Commercial - NoDerivatives 4.0"
Pas de réutilisation commerciale sans autorisation..
See: emh.ch/en/emh/rights-and-licences/
