Heutzutage liegt die Lebenserwartung von Patienten mit Zystischer Fibrose deutlich höher als noch vor wenigen Jahrzehnten – ein Grossteil der Betroffenen erreicht das mittlere Erwachsenenalter. Eine gut vorbereitete Transition in die Erwachsenenmedizin ist deshalb essentiell für die pädiatrischen Patienten.
Hintergrund
Zystische Fibrose (CF) ist eine der häufigsten autosomal-rezessiven Erbkrankheiten. In der Schweiz sind 800–1000 Menschen von dieser Multisystemkrankheit betroffen [1]. Aufgrund der CFTR-Genmutation («CF transmembrane conductance regulator»-Gen) sind die Chloridkanäle der exokrinen Drüsenzellen defekt, was eine zähe Schleimsekretion zur Folge hat. Noch vor wenigen Jahrzehnten starben die meisten CF-Patienten1 bereits im Kindes- und Jugendalter. Dank deutlich verbesserter symptomatischer Therapien und einer interdisziplinären und interprofessionellen Betreuung in CF-Zentren ist die Lebenserwartung heutzutage bis ins mittlere Erwachsenenalter gestiegen (2012 in Grossbritannien: 43,5 Jahre) [2]. Dies unterstreicht die Wichtigkeit einer strukturierten Transition. Generell ist die Transition definiert als geplanter Transfer von der Pädiatrie in die Erwachsenenmedizin bei Jugendlichen mit einer chronischen Krankheit wie zum Beispiel CF. Im Management von CF-Patienten hat die Transition eine enorme Bedeutung, ist zugleich aber eine grosse Herausforderung für Patienten, Angehörige und Behandlungsteams.
Das Leben eines Teenagers ist geprägt von Veränderungen in vielerlei Hinsicht: Abschluss der Schule und Einstieg ins Berufsleben, zunehmende Unabhängigkeit von den Eltern sowie die Entwicklung des eigenen Körperbilds. Eine Veränderung des medizinischen Settings ist in diesem Kontext eine zusätzliche Herausforderung. Der pädiatrische CF-Spezialist sowie alle anderen Mitglieder des interdisziplinären CF-Teams sind durch die langjährige Betreuung in der gesamten Kindheit oftmals zu Vertrauenspersonen geworden und es kann schwierig sein, diese in einer Zeit des generellen Umbruchs zu verlassen. Es ist deshalb ein grosser Schritt, im Zuge der Transition das eher geschützte und familienfokussierte pädiatrische Umfeld zu verlassen und in ein adultes Zentrum zu wechseln, wo beispielsweise die Autonomie und Selbstverantwortung des Patienten eine grössere Rolle spielen.
Zürcher Transitionsprogramm
In Zürich wurde vor über einem Jahrzehnt ein standardisiertes CF-Transitionsprogramm eingeführt. In halbjährlichen Abständen finden multidisziplinäre Transitions-Meetings statt, bei denen alle beteiligten Fachpersonen aus beiden CF-Zentren anwesend sind (Tab. 1). Alle wichtigen Aspekte zu den vor der Transition stehenden CF-Patienten werden ausführlich diskutiert. Alle Informationen werden in einem Transitionsbericht zusammengefasst und den Spezialisten am adulten CF-Zentrum übergeben. Dank dieser ausführlichen Übergabe wird eine zusätzliche Unruhe, wie sie zum Beispiel durch Anpassungen oder Umstellungen der medikamentösen Therapie entstehen würde, vermieden. Der letzte Schritt des Transitionsprozesses ist die Durchführung von zwei gemeinsamen Konsultationen des Patienten mit dem pädiatrischen sowie dem adulten CF-Spezialisten. Damit soll der Übergang bestmöglich gestaltet werden. Die Transition ist danach beendet, ein lockerer Austausch zwischen dem pädiatrischen und dem adulten CF-Zentrum besteht aber weiter. So wird an den oben erwähnten Transitionssitzungen häufig auch der weitere Verlauf bereits früher transitierter Patienten kurz diskutiert. Diese grossen Anstrengungen machen es den CF-Patienten möglich, sicher im neuem medizinischem Umfeld anzukommen.
Tabelle 1: Ablauf der standardisierten Transition von Patienten mit Zystischer Fibrose (CF) vom Kinderspital Zürich ans UniversitätsSpital Zürich.
Wer?
Was/Ziel?
1. Planung und Vorbereitung der Transition
Pädiatrisches CF-Team zusammen mit Patient und Angehörigen
Ansprechen und schrittweise Vorbereitung auf Transition, individuelle Planung des Transitionszeitpunktes, Erreichen einer stabilen Krankheitsphase
2. Multidisziplinäre Transitionssitzung
Jeweils aus beiden CF-Zentren: Ärzte, Pflegefachpersonen, Physiotherapeuten, Ernährungsberater, Sozialdienstmitarbeitende
Besprechung des einzelnen Patienten (medizinische Geschichte, familiäres und soziales Umfeld), angetroffene Schwierigkeiten im Patientenmanagement, Diskussion der individuellen Behandlungsstrategie für die Zukunft
3. Gemeinsame klinische Konsultationen
Patient + pädiatrischer CF-Spezialist + adulter CF-Spezialist (einmal am pädiatrischen, dann am adulten CF-Zentrum)
«Ablösung» vom pädiatrischen CF-Spezialisten, persönliche Übergabe und Vertrauensaufbau in zukünftigen adulten CF-Spezialisten
Zielsetzung und Methodik
Diese Studie wurde mit dem Ziel eines klinischen Audits durchgeführt, um diesen lange etablierten und standardisierten Transitionsprozess zwischen den beiden CF-Zentren (Kinderspital Zürich und UniversitätsSpital Zürich) zu evaluieren.
Alle CF-Patienten, die zwischen 07/2011 und 05/2014 über die reguläre Transition vom Kinderspital an das UniversitätsSpital Zürich wechselten, wurden in die Studie eingeschlossen. Eine retrospektive Datenanalyse aller konsekutiv transitierter CF-Patienten wurde durchgeführt. Primäre Endpunkte waren objektive klinische Parameter wie FEV1, Art und Anzahl der Atemwegspathogene und Body-Mass-Index (BMI) zum Zeitpunkt der Transition und ein Jahr später. Wir definierten die Transition als abgeschlossen nach Stattfinden der kombinierten klinischen Konsultation am adulten Zentrum. Es wurde deskriptive Statistik verwendet. Es wurde ein T-Test bei gepaarten Stichproben durchgeführt, eine Signifikanz akzeptiert bei p <0,05.
Bei der Ethikkommission des Kantons Zürich wurde vorgängig eine Bewilligung zur Durchführung der Studie eingeholt (angenommener Ethikantrag: KEK-ZH-Nr. 2015-0314). Einwilligungen aller Patienten zur anonymisierten Verwendung und Veröffentlichung der Daten liegen vor.
Wichtigste Ergebnisse
In der erwähnten Zeitperiode wurden 15 CF-Patienten regulär vom Kinderspital an das Universitätsspital Zürich transitiert (darunter vier Patientinnen). Die Genotypen der Studienpatienten entsprachen der CF-Genotypenverteilung in der Schweiz. Das mediane Alter zum Transitionszeitpunkt lag bei 18,9 Jahren (Range 16,8–21,8 Jahre). Sowohl im letzten Jahr vor als auch im ersten Jahr nach der Transition wurden die Patienten im Mittel fünfmal am jeweiligen CF-Zentrum gesehen. Alle Patienten konnten bis Studienende verfolgt werden.
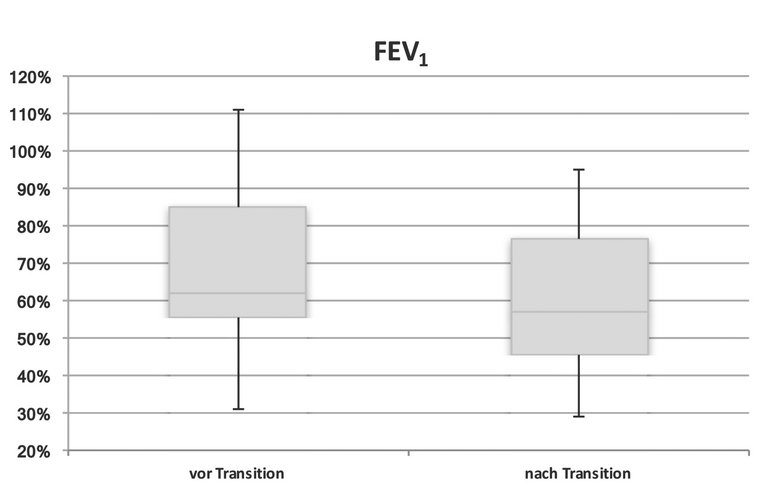
Art und Anzahl der Atemwegspathogene zum Zeitpunkt der Transition respektive ein Jahr danach blieben unverändert. Pseudomonas aeruginosa und Staphylococcus aureus waren die am häufigstem isolierten Sputumerreger. 36% der Patienten zeigten vor und nach der Transition einen chronischen pulmonalen Infekt mit Pseudomonas aeruginosa. Dieser Befund geht mit einem negativen Effekt auf die Lungenfunktion einher und ist oft mit niedrigeren FEV1-Werten assoziiert [3]. Bei jedem Patienten wurde der Durchschnitt der letzten drei Spirometrieresultate vor der Transition beziehungsweise von drei Spirometriemessungen ein Jahr nach Transition berechnet. Der mediane FEV1-Sollwert lag in der Patientengruppe vor der Transition bei 62% (Range 31–111%) und nach der Transition bei 57% (29–95%), p = 0,01, siehe Abbildung 1. Zwei Patienten zeigten nach Transition einen höheren FEV1-Wert, vier Patienten hatten nach Transition einen FEV1-Verlust von durchschnittlich 17,5% (16–20%) zu verzeichnen. Alle fünf mit Pseudomonas aeruginosa chronisch infizierten Patienten waren unter inhalativer antibiotischer Dauertherapie (Monotherapie mit Tobramycin im monatlichen «on/off»-Schema oder Kombinationstherapie von Tobramycin mit Colistin oder Aztreonam). Vor der Transition befanden sich 80% der Patienten in regelmässiger physiotherapeutischer Behandlung, nach Transition in die Erwachsenenmedizin 53%.
Abbildung 1: FEV 1 vor beziehungsweise nach Transition (p <0,05).
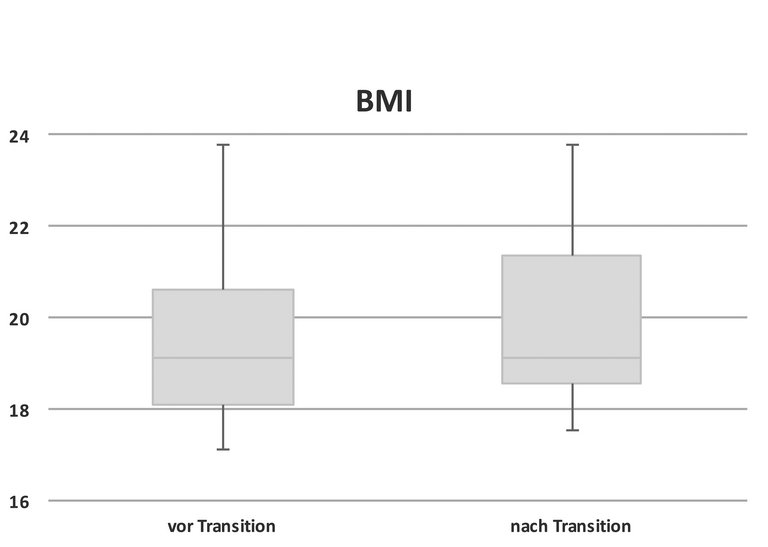
Bis auf einen Studienpatienten konnten alle ihren BMI während der Transition stabil halten oder sogar erhöhen (BMI vor und nach Transition im Median 19,1 kg/m2; Range vor Transition 14,5–23,8 kg/m2 bzw. danach 15,7–23,8 kg/m2, p = 0,44, siehe Abb. 2). Die Messung und Dokumentation des BMI ist ein wichtiger Bestandteil in der Betreuung von CF-Patienten, die Korrelation des Ernährungszustandes mit der Lungenfunktion wurde mehrfach gezeigt [4–6].
Abbildung 2: Body-Mass-Index (BMI) vor beziehungsweise nach Transition (p >0,05).
Ein Patient hatte vor der Transition einen insulinpflichtigen CF-assoziierten Diabetes mellitus; innerhalb eines Jahres nach der Transition wurde bei zwei weiteren Patienten ein insulinpflichtiger Diabetes mellitus diagnostiziert. Zur Bestimmung der Knochendichte wurden DEXA-Messungen durchgeführt; bei zwei Patienten wurde eine zu tiefe Knochendichte nachgewiesen (Z-Score Lendenwirbelsäule ≤–2,0).
Schlussfolgerungen und Ausblick
In Zürich besteht ein standardisiertes Transitionsprogramm mit multidisziplinären Besprechungen und gemeinsamen klinischen Konsultationen zwischen dem pädiatrischen CF-Team und dem Team am adulten CF-Zentrum. Unsere Daten zeigen, dass das etablierte Programm gut funktioniert und die kontinuierliche spezialisierte CF-Betreuung garantiert ist. Die Patienten wiesen auch nach Transition insgesamt einen stabilen Gesundheitszustand auf. Der signifikante FEV1-Unterschied von 5% ist höher als der aufgrund der Grunderkrankung CF zu erwartende jährliche FEV1-Verlust, verursacht bei kleiner Fallzahl durch die vier (oben erwähnten) Patienten mit einem überdurchschnittlichen FEV1-Verlust. Diese Verluste sind am ehesten einer suboptimalen Therapieadhärenz während des Transitionsprozesses zuzuschreiben. Bei allen vier Patienten zeigten sich jedoch erfreuliche klinische und lungenfunktionelle Verläufe (mit verbesserten oder mindestens stabilen FEV1-Werten) bis heute.
Die Transition ist ein schrittweiser Prozess und soll durch den Pädiater schon früh angesprochen werden, um den Patienten bestmöglich vorzubereiten (Therapieadhärenz und Selbstmanagement des Patienten sind wichtige Grundvoraussetzungen für eine erfolgreiche Transition). Der Zeitpunkt der Transition muss individuell in Bezug auf das chronologische Alter, die physische und kognitive Reife des Patienten erfolgen [7]. Weiter soll die Transition in einer möglichst stabilen Krankheitsphase sowie zu einem günstigen Zeitpunkt in Bezug auf die Ausbildung (z.B. nach Lehrabschluss oder nach Matura) erfolgen. Vor, während und kurz nach dem Transitionsprozess erachten wir das sorgfältige Monitoring der Patienten in regelmässigen Intervallen sowie der Hinweis auf die Bedeutung von täglich durchgeführter Therapie für einen adäquaten Gesundheitszustand der CF-Patienten als besonders entscheidend. Das Motivieren für den Besuch regelmässiger Physiotherapie stellte beispielsweise in dieser Patientengruppe eine grosse Herausforderung dar. Das Thema der Adhärenz bei Jugendlichen und jungen Erwachsenen im Transitionsprozess bleibt auch in Zukunft ein Fokus.
Eine enge Zusammenarbeit zwischen pädiatrischem und adultem CF-Zentrum ermöglicht eine kontinuierliche, dem Patienten angepasste Betreuung. Auch in Zürich kann diese Zusammenarbeit zwischen den beiden CF-Zentren noch weiter intensiviert werden.
In Zukunft erscheint die Einführung eines national standardisierten und in Kooperation mit der «Swiss Working Group for Cystic Fibrosis» (SWGCF) erarbeiteten CF-Transitionsprogramms begrüssenswert.
Eine optimal terminierte und gut vorbereitete Transition in die Erwachsenenmedizin ist für alle pädiatrischen Patienten mit chronischen Krankheiten von grosser Bedeutung [8]. Wir sind deshalb der Überzeugung, dass unser gut etabliertes Zürcher CF-Transitionsprogramm ein gutes Modell auch für die Transition vieler anderer chronisch kranker Jugendlicher bieten kann.
Disclosure statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Correspondance
cand. med. Simon Klöti Klinik für Pneumologie UniversitätsSpital Zürich Rämistrasse 100 CH-8091 Zürich simon.kloeti[at]uzh.ch
PD Dr. med. Christian Benden Klinik für Pneumologie UniversitätsSpital Zürich Rämistrasse 100 CH-8091 Zürich christian.benden[at]usz.ch
Literatur
1 www.cfch.ch (Schweizerische Gesellschaft für Cystische Fibrose)
2 UK Cystic Fibrosis Registry, Annual Data Report 2013. 2014.
3 Kerem E, Viviani L, Zolin A, MacNeill S, Hatziagorou E, Ellemunter H, et al. Factors associated with FEV1 decline in cystic fibrosis: analysis of the ECFS patient registry. Eur Respir J. 2014;43(1):125–33.
5 Milla CE. Nutrition and lung disease in cystic fibrosis. Clin Chest Med. 2007;28(2):319–30.
6 Stallings VA, Stark LJ, Robinson KA, Feranchak AP, Quinton H, Subcommittee CPGoGaN, et al. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc. 2008;108(5):832–9.
7 LaRosa C, Glah C, Baluarte HJ, Meyers KE. Solid-organ transplantation in childhood: transitioning to adult health care. Pediatrics. 2011;127(4):742–53.
8 Greenlee MC, D’Angelo L, Harms SR, Kuo AA, Landry M, McManus M, et al. Enhancing the Role of Internists in the Transition From Pediatric to Adult Health Care. Ann Intern Med. 2017;166(4):299–300.