La sclérose systémique est certes une connectivite rare, mais son évolution est potentiellement grave, et cette affection s’accompagne souvent d’une morbidité considérable et d’une mortalité accrue. Une guérison n’est pas en vue; dans de nombreux cas, seul un traitement de soutien est possible. Cela peut provoquer une incertitude et une anxiété chez le patient et une certaine impuissance chez le médecin. Cet article a pour objectif de fournir au praticien quelques conseils pour la pose du diagnostic, de présenter des propositions de traitement pour les symptômes les plus fréquents et d’encourager la prise en charge thérapeutique interdisciplinaire.
Introduction
La sclérose systémique est une connectivite rare caractérisée par une vasculopathie, une fibrose et une auto-immunité. Elle se manifeste plus fréquemment chez les femmes que chez les hommes (4:1), généralement à l’âge de 35–65 ans. Toutefois, toutes les tranches d’âge peuvent être touchées [1].
En fonction de l’étendue de l’atteinte cutanée, la distinction est faite entre une forme limitée et une forme diffuse. Les manifestations au niveau des organes peuvent survenir dans les deux formes, mais avec une prévalence différente. Elles peuvent concerner le tractus gastro-intestinal, les poumons, les reins, le système musculo-squelettique et le cœur. Il convient d’éviter d’employer le terme «syndrome CREST», car les manifestations CREST appartiennent à la forme limitée. En outre, ce terme pourrait donner l’impression d’une évolution bénigne, alors que les patients peuvent non seulement développer les manifestations pathologiques regroupées sous l’acronyme CREST (calcinose, phénomène de Raynaud, trouble de la motilité œosophagienne, sclérodactylie, télangiectasies), mais également d’autres complications graves, telles que des ulcérations digitales ou une hypertension artérielle pulmonaire.
Le traitement dépend de l’organe atteint. Bien qu’il n’existe aucun traitement de fond pour la sclérose systémique, d’importants progrès ont été réalisés au cours des dernières années, avant tout en ce qui concerne le traitement de l’hypertension artérielle pulmonaire. Par ailleurs, il existe de nouveaux principes actifs prometteurs contre la pneumopathie interstitielle, qui font actuellement l’objet d’études.
L’inclusion des patients dans l’EUSTAR («European Scleroderma Trials and Research Group»), une cohorte européenne de patients atteints de sclérose systémique, permet d’acquérir des informations scientifiques précieuses à propos de cette maladie rare.
Nouveaux critères de classification
Les nouveaux critères de classification ACR/EULAR1 (2013) ont remplacé les anciens critères de classification ARA2 (1980) et disposent d’une meilleure sensibilité et d’une meilleure spécificité (tab. 1). Il ne faut toutefois pas oublier que ces critères ne servent pas à poser le diagnostic, mais ont pour principale vocation de servir la recherche. Les formes précoces de la maladie peuvent également être «manquées» avec l’utilisation des nouveaux critères. En revanche, une atteinte cutanée étendue permet déjà de poser le diagnostic de sclérose systémique. Désormais, d’autres manifestations de la maladie trouvent également leur place parmi les nouveaux critères de classification, qu’elles soient cliniques (phénomène de Raynaud, tuméfaction des doigts) ou paracliniques (anticorps spécifiques, hypertension artérielle pulmonaire ou capillaroscopie pathologique). En particulier le phénomène de Raynaud, la tuméfaction des doigts ainsi que des anticorps antinucléaires (ANA) positifs peuvent constituer des signes subtils d’une sclérose systémique débutante. L’orientation vers un spécialiste pour des examens complémentaires est déjà indiquée à ce stade, car un traitement à un stade précoce de la maladie s’accompagne d’un meilleur pronostic (tab. 1).
Tableau 1: Critères de classification.
Critères de classification ACR/EULAR 2013 Un total ≥9 points permet de diagnostiquer une sclérose systémique.
Critères
Sous-critères
Points
Epaississement de la peau des doigts au niveau des deux mains. Atteinte proximale au-delà de la région des articulations de la base des doigts (critère suffisant)
−
9
Epaississement de la peau des doigts
Doigts tuméfiés
2
Sclérodactylie
4
Lésions de la pointe des doigts
Ulcères
2
Cicatrices en forme de fossettes
3
Télangiectasie
–
2
Capillaires anormaux de la base de l’ongle
–
2
Hypertension artérielle pulmonaire ou pneumo- pathie interstitielle (maximum 2 points)
Hypertension artérielle pulmonaire
2
Pneumopathie interstitielle
2
Phénomène de Raynaud
–
3
Auto-anticorps associés à la SSc (anti-centromères, anti-Scl-70, anti-ARN-polymérase III) (maximum 3 points)
Anti-centromères
3
Anti-Scl-70
3
Anti-ARN-polymérase III
3
ACR/EULAR = American College of Rheumatology / European League Against Rheumatism
Manifestations cliniques
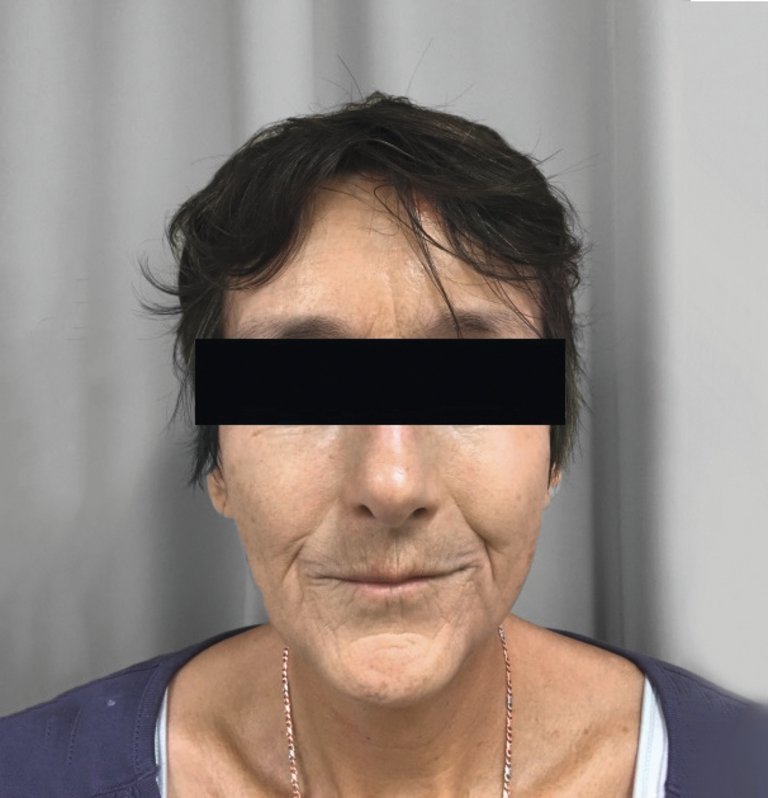
En cas de sclérose systémique limitée, les altérations cutanées sont distales par rapport aux coudes et aux genoux. Pour la forme diffuse, celles-ci se trouvent également dans les zones proximales, ainsi que sur le tronc. Au niveau du visage, une réduction de l’ouverture de la bouche d’aspect typique et une perte de l’expression faciale sont possibles dans les deux formes de la maladie (fig. 1).
Figure 1: Patiente de 55 ans atteinte de sclérose systémique limitée depuis 8 ans, avec bouche présentant un aspect typique (la publication a été réalisée avec l’accord de la patiente).
Initialement, il existe le plus souvent une prolifération inflammatoire des fibres de collagène avec formation d’œdème; plus tard survient une fibrose avec durcissement de la peau. Au niveau des doigts, on trouve souvent un flexum avec perte secondaire de la fonction de préhension (fig. 2). Il n’est pas rare que la peau ne soit pas du tout touchée; on parle alors de «sclérose systémique sine scleroderma».
Figure 2 A et B: Fibrose cutanée avec flexum secondaire et ulcérations des doigts (flèches dans A) chez un patient atteint de sclérose systémique diffuse.
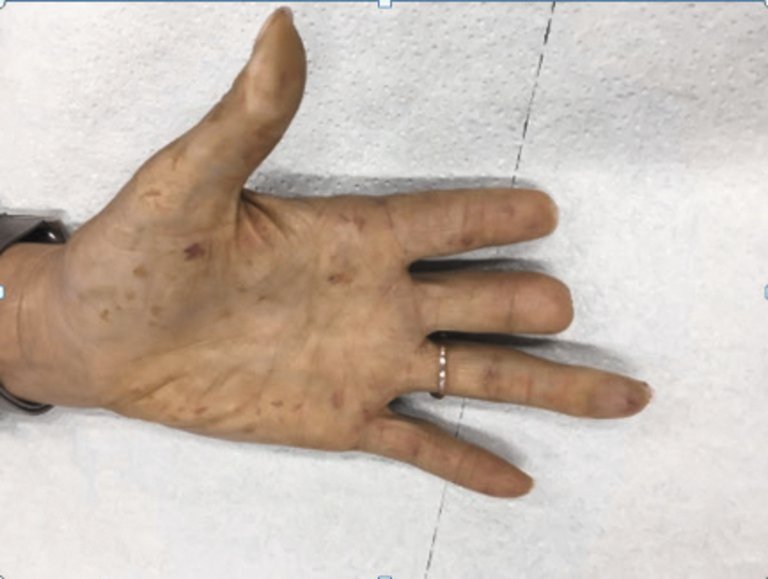
Le phénomène de Raynaud (fig. 3) est la première manifestation de la maladie. Il est présent chez plus de 85% des patients, évolue souvent de façon croissante, est responsable d’une ischémie et entraîne une nécrose acrale (ulcère du bout des doigts) dans jusqu’à 50% des cas (fig. 4). Dans la forme limitée, le phénomène de Raynaud survient souvent longtemps avant les autres manifestations de la maladie. Les altérations cutanées se développent habituellement lentement. Une hypertension artérielle pulmonaire peut se manifester des années plus tard en tant qu’expression supplémentaire de la vasculopathie. Dans la forme diffuse, le phénomène de Raynaud se manifeste souvent peu de temps avant ou en même que les altérations cutanées. Au fil du temps, les personnes touchées développent rapidement des manifestations viscérales telles que la pneumopathie interstitielle («interstitial lung disease» [ILD]) ou la crise rénale.
Figure 3: Phénomène de Raynaud. Chez cette patiente de 33 ans, on remarque une coloration pâle de l’annulaire droit (flèche) avec une coloration diffuse et légèrement violette des autres doigts et des paumes des mains.
Figure 4: Chez cette patiente, l’ischémie s’est manifestée sous forme extrême par des nécroses des phalanges distales de l’index et du majeur qui, malgré un traitement vasodilatateur, ont nécessité une amputation. La patiente présente également des télangiectasies typiques sur tout l’intégument et au niveau des muqueuses, bien visibles ici au niveau de la paume (la publication a été réalisée avec l’accord de la patiente.).
Les arthrites ou arthralgies sont relativement fréquentes et affectent principalement les petites articulations. A la différence de l’arthrite rhumatoïde, l’évolution est généralement bénigne et non érosive.
Pour les deux formes de la maladie, la plupart des patients présentent une dysphagie et/ou des problèmes de reflux relativement tôt après le début de la maladie, et sont l’expression de la dysmotilité œsophagienne. Ils peuvent développer un trouble de la motilité étendu à l’ensemble du tractus gastro-intestinal, qui peut conduire à une parésie intestinale avec malabsorption et malnutrition, associée à une réduction de la qualité de vie. Les sténoses œsophagiennes sont une autre complication tardive.
La survenue d’une arythmie, d’une dysfonction diastolique ou d’une insuffisance cardiaque peut indiquer une atteinte cardiaque. Des épanchements péricardiques, généralement hémodynamiquement neutres, sont aussi souvent présents. Les grands épanchements péricardiques sont toutefois associés à une hypertension artérielle pulmonaire et à un pronostic défavorable.
Une complication redoutée de la sclérose systémique diffuse est la crise rénale. Avant l’ère des inhibiteurs de l’enzyme de conversion de l’angiotensine (IEC), l’atteinte rénale était la principale cause de mortalité. Même en cas d’initiation précoce d’un traitement adéquat, le pronostic de survie reste aujourd’hui médiocre. Les signes d’alerte d’une possible atteinte rénale sont une augmentation de la pression artérielle (>30 mm Hg systolique ou >20 mm Hg diastolique), une dégradation de la fonction rénale ou un sédiment urinaire actif. Il a été clairement montré qu’un traitement par glucocorticoïdes peut favoriser la crise rénale (avant tout en cas de doses ≥15 mg d’équivalent prednisolone); il est dès lors nécessaire d’y renoncer autant que possible.
Diagnostic
Des ANA positifs sont détectés dans le sérum dans plus de 95% des cas. Parmi les différents anticorps habituellement déterminés dans le cadre de l’évaluation initiale, nombreux sont ceux qui montrent une association avec certaines caractéristiques de la maladie. Les anticorps anti-centromères sont par exemple associés à l’atteinte cutanée limitée et à l’hypertension artérielle pulmonaire; en revanche, les patients touchés ont un faible risque de développer une pneumopathie interstitielle. Les anticorps anti-Scl-70 sont quant à eux associés à l’atteinte cutanée diffuse et à la fibrose pulmonaire. Les anticorps anti-RNA polymérase III prédisposent à la crise rénale.
La capillaroscopie est une méthode sûre et non invasive pour évaluer la microvasculopathie au niveau de la base de l’ongle. Elle permet le plus souvent de faire la différence entre un phénomène de Raynaud primaire et secondaire. Trois stades typiques d’altérations microvasculaires sont décrits en cas de sclérose systémique:
– «early», avec quelques mégacapillaires et peu d’hémorragies (fig. 5);
Figure 5: Vidéocapillaroscopie A) chez un sujet sain avec aspect «en palissade» des capillaires, qui apparaissent généralement en forme d’épingle à cheveux; B) chez une patiente atteinte de sclérose systémique limitée, avec de nombreux mégacapillaires.
– «active», avec de nombreux mégacapillaires, de nombreuses hémorragies et une raréfaction capillaire débutante;
– «late», avec une raréfaction capillaire étendue.
Il est intéressant de constater que les anticorps anti-centromères sont en corrélation avec le type «active», et les anticorps anti-Scl-70 avec le type «late»[2]. Une densité capillaire réduite est un facteur prédictif positif de récidive d’ulcères des doigts. Dans ce contexte, il convient toutefois de mentionner que les antécédents d’ulcères ou les ulcères actifs représentent le principal facteur de risque d’une telle récidive. Il peut également y avoir des altérations pathologiques à la capillaroscopie dans d’autres connectivites (typiquement en cas de dermatomyosite, collagénose mixte et plus rarement en cas de lupus érythémateux systémique); le diagnostic de la sclérose systémique doit dès lors s’effectuer en tenant compte de l’ensemble des éléments cliniques.
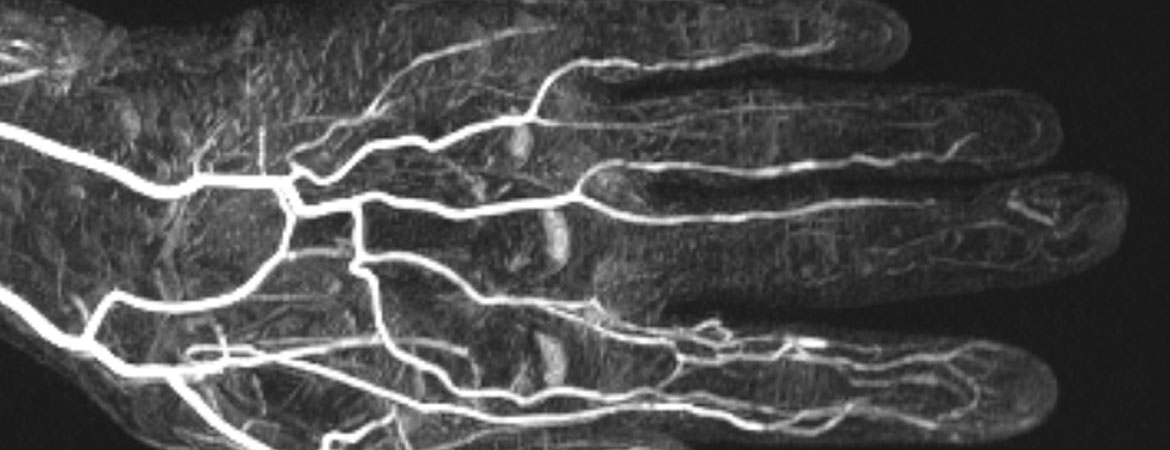
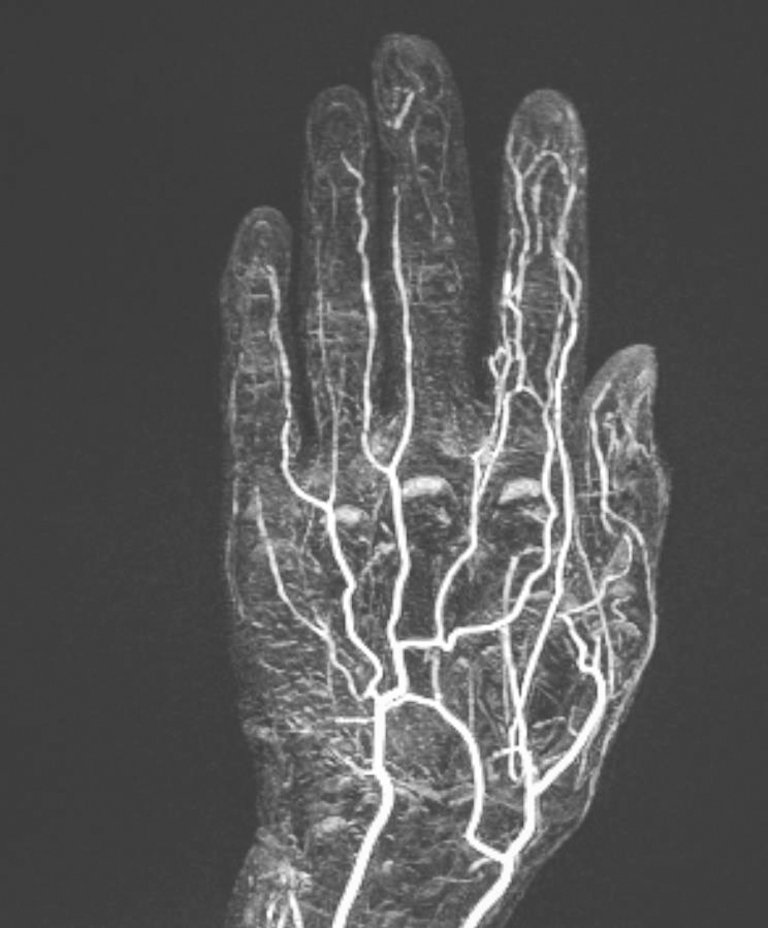
En cas d’ischémie acrale d’origine indéterminée, la réalisation d’une angiographie par résonance magnétique peut également aider à clarifier l’étiologie. Celle-ci reflète la vasculopathie des artères digitales, souvent accompagnée d’une occlusion distale (fig. 6).
Figure 6: L’angiographie par résonance magnétique de la main gauche montre un contraste réduit des artères digitales des rayons III–V chez un patient présentant un ulcère du majeur.
Une «banale» radiographie des mains permet également parfois de confirmer le diagnostic de sclérose systémique, au cas où elle révèle une calcinose ou une ostéolyse acrale, comme l’illustre la figure 7.
Figure 7: Calcinose cutanée. A) Radiographie de la main gauche, calcinose le mieux visible au niveau du rayon I et V; B) échographie du pouce, palmaire, chez la même patiente: la calcinose se reconnait par sa structure hyperéchogène (flèche) en forme de nuages, avec cône d’ombre postérieur (étoile).
Initialement, tous les patients doivent bénéficier d’un examen complet de la fonction pulmonaire (comprenant une mesure de la capacité de diffusion du monoxyde de carbone). En fonction de l’évolution, des contrôles devraient être réalisés tous les 3 à 12 mois. Le CT-scanner thoracique avec coupes fines présente une bien meilleure sensibilité pour les altérations pulmonaires interstitielles par rapport à la radiographie conventionnelle; il représente dès lors l’examen de choix. Comme observation secondaire, le CT-scanner montre souvent une dilatation de l’œsophage, signe évident d’un trouble de sa motilité. Toutefois, ce trouble est habituellement recherché au moyen d’un transit baryté ou d’une manométrie-impédance. Bien que cette dernière soit plus invasive, elle présente une bien meilleure sensibilité et spécificité.
L’échocardiographie peut montrer des signes indirects d’une hypertension artérielle pulmonaire; pour confirmation, il est toutefois nécessaire de réaliser un cathétérisme cardiaque droit.
Stratégies thérapeutiques
Dans la mesure où il n’existe aucun traitement de fond de la sclérose systémique, un traitement spécifique à l’organe touché n’est initié que lorsqu’un problème spécifique se manifeste (tab. 2). La difficulté réside dans la détection précoce du problème, ce qui implique une collaboration étroite entre les médecins traitants et une information et formation approfondies du patient.
Soins de la peau, physiothérapie (pour le drainage lymphatique, l’amélioration de l’élasticité cutanée, la prévention/le traitement des contractures articulaires secondaires)
Immunosuppresseurs (administration hebdomadaire de méthotrexate 15 mg par voie sous-cutanée, cyclophosphamide par voie intraveineuse en «pulse therapy»)
Antagonistes des récepteurs de l’endothéline (par ex. Opsumit 10 mg 1×/jour, Tracleer 125 mg 2×/jour, Volibris 5–10 mg 1×/jour)
Inhibiteurs de la PDE5 (par ex. Sildenafil 20–50 mg 3×/jour, Tadalafil 40 mg 1×/jour)
Riociguat (Adempas) 1–2,5 mg 3×/jour
Analogues de la prostacycline (par ex. Ilomedin par voie intraveineuse en perfusion continue ou Remodulin par voie intraveineuse ou sous-cutanée en perfusion continue)
Selexipag (Uptravi 200–1600 mcg 2×/jour)
Pneumopathie interstitielle
Immunosuppression (cyclophosphamide par voie intraveineuse en «pulse therapy», mycophénolate mofétil)
Crise rénale
Inhibiteurs de l’enzyme de conversion de l’angiotensine, le cas échéant, antagonistes des récepteurs de l’angiotensine et autres antihypertenseurs
Le cas échéant, dialyse
Forme sévère de progression rapide
Transplantation autologue de cellules souches
* nécessite une garantie de prise en charge des coûts pour cette indication
En particulier en cas de forme diffuse, des évaluations trimestrielles doivent initialement être réalisées par l’équipe professionnelle. En cas d’évolution stable et en particulier en cas de forme limitée, des contrôles annuels peuvent éventuellement suffire.
Pour lutter contre la sclérose cutanée et la vasculopathie digitale, il est en première ligne recommandé d’instaurer des mesures non médicamenteuses, telles que les bains de paraffine, l’arrêt du tabagisme et la prévention des traumatismes. En ce qui concerne le phénomène de Raynaud, la protection contre le froid est absolument essentielle; le patient devrait garder au chaud l’intégralité de son corps, et pas uniquement ses membres. Dans notre clinique, les personnes touchées reçoivent des conseils relatifs aux mesures de protection contre le froid dans le cadre d’une consultation spéciale en ergothérapie. En outre, des inhibiteurs calciques ou des préparations à base de nitrate en application locale peuvent être utilisés. Les inhibiteurs de la phosphodiestérase de type 5 (PDE5) représentent également une option, mais ne sont pas admis en Suisse par les caisses-maladie pour cette indication. En raison d’interactions, la combinaison avec des préparations à base de nitrate (que ce soit en application orale ou locale) est interdite. Selon les nouvelles recommandations, l’utilisation de fluoxétine peut également être tentée. L’administration intraveineuse d’iloprost (analogue de la prostacycline) est très efficace contre les ulcères des doigts ou l’ischémie acrale critique (fig. 8). En cas d’effet insuffisant, le sildénafil (inhibiteur de la PDE5) peut être prescrit en plus, mais nécessite une garantie de prise en charge des coûts. En Suisse, seul le bosentan (antagoniste des récepteurs de l’endothéline) est admis par les caisses-maladie pour la prophylaxie des récidives. Le bosentan est utilisé en particulier chez les patients présentant de multiples ulcères malgré un traitement conventionnel, et ne contribue pas à la guérison des ulcères.
Figure 8: Evolution après traitement vasodilatateur par illoprost.
En cas d’atteinte cutanée, le soin de la peau est essentiel; le traitement physio- et ergothérapeutique revêt quant à lui une importance majeure en cas de problématique secondaire avec œdème ou tendance aux contractures articulaires. Il n’est pas rare que les personnes touchées requièrent un traitement régulier (souvent hebdomadaire) pour le maintien d’une mobilité aussi bonne que possible, en particulier des articulations de la main et des doigts. Des exercices d’étirement réalisés par le patient lui-même peuvent également contribuer à l’amélioration de la mobilité. Avec le temps, des patients peuvent présenter une amélioration spontanée des altérations cutanées. Toutefois, il est fréquent que les contractures déjà présentes persistent. L’utilisation de méthotrexate est recommandée en cas de sclérose systémique diffuse de stade précoce. En cas d’évolution rapidement progressive, il convient également d’envisager l’administration de cyclophosphamide, malgré la toxicité élevée.
Des mesures locales sont indiquées pour lutter contre le syndrome sec qui peut, comme en cas de syndrome de Sjögren primaire, être très invalidant. La pilocarpine en gouttes ou en comprimés (Salagen®) peut également être utile pour la xérophtalmie. Il convient de veiller à une bonne hygiène dentaire. Souvent, la caisse-maladie prend en charge une partie du traitement d’hygiène dentaire, sur demande.
En cas d’atteinte gastro-intestinale, des prokinétiques peuvent être utilisés à un stade précoce; en revanche, dans les stades avancés, ceux-ci n’agissent plus en raison de la fibrose croissante. En principe, tous les patients devraient recevoir un traitement par inhibiteurs de la pompe à protons (IPP), car ceux-ci permettent de prévenir le reflux gastro-œsophagien, les sténoses œsophagiennes distales secondaires et une dilatation par la bougie de ces dernières. En cas de diarrhée récidivante et dans l’hypothèse d’une prolifération bactérienne, il est possible d’initier un traitement antibiotique correspondant. En présence de signes d’une malabsorption sévère résistante au traitement accompagnée de cachexie, il convient d’envisager la mise en place d’une nutrition parentérale; celle-ci peut être initiée en milieu hospitalier puis poursuivie à domicile sous surveillance régulière. Les patients gagnent ainsi en espérance de vie et en qualité de vie.
De nombreux progrès ont été accomplis au cours des dernières années dans le domaine du traitement de l’atteinte pulmonaire, que ce soit pour l’hypertension artérielle pulmonaire ou la pneumopathie interstitielle.
En cas d’hypertension artérielle pulmonaire, il convient de souligner qu’un diagnostic précoce est bénéfique pour le pronostic même chez les patients asymptomatiques. Cela souligne l’importance du dépistage régulier de l’hypertension artérielle pulmonaire. Jusqu’à présent, les antagonistes des récepteurs de l’endothéline et les inhibiteurs de la PDE étaient utilisés en traitement de première ligne, avec en plus un analogue de la prostacycline administré par voie parentérale pour les cas graves. Depuis 2 ans, nous disposons également du riociguat (un stimulateur de la guanylate cyclase) pour cette indication. Le sélexipag, le premier agoniste des récepteurs de la prostacycline, est nouvellement autorisé en Suisse. Par rapport aux analogues de la prostacycline, le sélexipag présente le grand avantage d’une administration orale; l’augmentation progressive de la dose requiert toutefois une surveillance régulière et une information détaillée du patient [4]. De récentes études montrent qu’un traitement d’association est probablement supérieur à la monothérapie, et ce dès les stades précoces. En cas d’hypertension artérielle pulmonaire, la décision thérapeutique incombe idéalement à une équipe interdisciplinaire composée de rhumatologues, pneumologues et cardiologues.
Dans le cas de la pneumopathie interstitielle, il convient d’utiliser en première ligne le cyclophosphamide. Grâce à de récentes données, il est désormais possible d’envisager l’administration de mycophénolate mofétil. Dans une étude récemment publiée, l’effet thérapeutique a été jugé comparable à celui du cyclophosphamide. Le profil d’effets indésirables penchait toutefois en faveur du mycophénolate mofétil. La pirfénidone, un principe actif antifibrotique déjà utilisé dans le cadre de la fibrose pulmonaire idiopathique, a récemment été évalué quant à sa tolérance chez les patients atteints de sclérose systémique. Les données n’ont toutefois pas permis de tirer de conclusions quant à l’effet thérapeutique. Etant donné que le reflux gastro-œsophagien a une influence négative sur la pneumopathie interstitielle, un traitement par IPP est impératif en cas de pneumopathie interstitielle.
La crise rénale constitue une urgence; son traitement doit être initié rapidement en milieu hospitalier. Les IEC représentent la première option thérapeutique, dans la mesure où le taux de survie en cas d’atteinte rénale s’est massivement améliorer depuis leur introduction. Il est par conséquent recommandé de privilégier les IEC, même en présence de signe d’une hypertension artérielle seulement légère. Parfois, malgré tous les efforts, le patient nécessite d’être dialysé.
En cas de maladie rapidement progressive, il est possible d’envisager, dans des cas spécifiques, une transplantation de cellules souches (tab. 2).
Pronostic
Le pronostic est très variable. La forme limitée s’accompagne en principe d’un meilleur pronostic. La présence d’une atteinte organique (poumon, rein, cœur) et le sexe masculin sont associés à un moins bon pronostic.
Perspectives
Les antifibrotiques sont plus que jamais nécessaires. Le nintédanib (inhibiteur de la tyrosine kinase) et la pirfénidone sont utilisés en cas de fibrose pulmonaire idiopathique et semblent aussi très prometteurs pour le traitement de la pneumopathie interstitielle dans le cadre de la sclérose systémique. Dans un collectif de patients présentant des paramètres inflammatoires élevés, le tocilizumab a montré une tendance à l’amélioration des altérations cutanées. De la même manière, dans de petites études, le rituximab a entraîné une légère amélioration des altérations cutanées, ainsi qu’une légère amélioration et stabilisation des fonctions pulmonaires. D’autres études évaluant l’effet thérapeutique de ces substances en cas de sclérose systémique sont prévues ou d’ores et déjà en cours; leurs résultats devraient être publiés au cours des mois ou années à venir [5].
L’essentiel pour la pratique
• En cas de phénomène de Raynaud d’apparition nouvelle ou se dégradant, ainsi qu’en cas de tuméfaction des doigts inexpliquée, il est judicieux de déterminer le titre d’anticorps antinucléaires (ANA) et d’orienter les patients afin de mener d’autres investigations.
• Un contrôle régulier (initialement mensuel) du statut urinaire et de la pression artérielle par les patients eux-mêmes et le médecin de famille est essentiel. En cas de suspicion de crise rénale, le patient doit être immédiatement adressé à un spécialiste.
• Le dépistage de l’hypertension artérielle pulmonaire devrait être réalisé de façon conséquente, car un traitement précoce est associé à un meilleur pronostic. Un traitement d’association doit entrer tôt en considération.
• Le sexe masculin, la forme diffuse et la présence d’une atteinte organique sont associés à un pronostic défavorable.
• Une surveillance régulière (au moins annuelle) par une équipe de spécialistes expérimentés est recommandée, même chez les patients asymptomatiques, afin de garantir l’initiation d’un traitement en temps voulu.
Remerciements
L’auteure remercie chaleureusement le Professeur Peter Villiger, de la clinique de rhumatologie, immunologie et allergologie de l’hôpital universitaire de Berne, ainsi que le Docteur Magdalena Gantenbein, spécialiste en médecine interne générale à Bâle, pour la relecture critique du manuscrit et les suggestions constructives.
Disclosure statement
L’auteure n’a pas déclaré d’obligations financières ou personnelles en rapport avec l’article soumis.
Correspondance
Dr méd. Diana Dan Service de Rhumatologie Hôpital orthopédique CHUV Avenue Pierre-Decker CH-1011 Lausanne diana.dan-rusei[at]chuv.ch
Références
1 Dan D. Die Systemsklerose. Der informierte Arzt. 2012;2:14–5.
2 Cutolo M, Editor. Atlas of capillaroscopy in rheumatic diseases. 1st ed. Milano: Elsevier;2010.
3 Villiger P. Systemsklerose. Therapeutische Umschau. 2008;65:283–7.
4 Kowal-Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2016;0:1–13.
5 Khanna D, Distler JHW, Sandner P, Distler O. Emerging strategies for treatment of systemic sclerosis. Journal of Scleroderma and Related Disorders. 2016;1(2):186–93.