a Klinik für Medizinische Hämatologie und Onkologie, Stadtspital Triemli, Zürich; b Klinik für Neurologie, Stadtspital Triemli, Zürich; c Medizinische Klinik und Poliklinik (Onkologie, Hämatologie und Knochenmarkstransplantation mit Sektion Pneumologie), Universitätsklinikum Hamburg-Eppendorf, Deutschland
Die Assoziation der Myasthenia gravis mit Thymomen ist gut bekannt. Selten koexistiert eine Myasthenia gravis mit einem Lymphom. Bei unklaren neurologischen Symptomen, welche durch die klinische Untersuchung und Bildgebung nicht abschliessend erklärt werden können, müssen paraneoplastische Syndrome in Erwägung gezogen werden.
Fallbericht
Anamnese
Die notfallmässige Hospitalisation des 80-jährigen Patienten erfolgte bei Allgemeinzustandsverschlechterung mit Inappetenz und Gewichtsverlust von 15 kg. Fieber und Nachtschweiss wurden verneint. Im stationären Verlauf jedoch beschwerte sich der Patient über zunehmende Schluckbeschwerden mit Aspirationen.
Der bis vor kurzem noch täglich handwerklich tätige Patient zeigte sich stationär zunehmend immobil sowie bettlägerig und empfand neben einer generalisierten Schwäche ebenfalls zunehmend Schwierigkeiten, sich verbal auszudrücken. In der ergänzenden Systemanamnese berichtet der Patient von einer Anstrengungsdyspnoe (NYHA II–III) sowie einer Obstipationsneigung.
Die Vorgeschichte war bis auf eine medikamentös behandelte arterielle Hypertonie bland.
Status (nach erfolgter Lymphomdiagnose)
Der Patient präsentierte sich in reduziertem Allgemeinzustand und kompensiertem Ernährungszustand (BMI 22 kg/m2). Blutdruck 150/91 mm Hg, Puls 102/min, afebril, SaO2 92% bei Raumluft. Enoral ausgeprägte Exsikkose mit verschorftem Gaumensegel beidseits. Vergrösserte und indolente Lymphknoten submandibulär und supraklavikulär beidseits. Pulmonal reduziertes Atemgeräusch linksbasal. Abdominal diffuse Druckdolenz allseits sowie Splenomegalie. Neurologisch wach, voll orientiert. Leichte Ptose beidseits ohne Zunahme im Simpson-Test, keine Doppelbilder, Fazialis seitengleich, hypophone Dysarthrie, Zunge mittig. Armvorhalteversuch über eine Minute ohne Absinken, Beinvorhalteversuch nur über wenige Sekunden möglich. Keine höhergradigen Paresen. Muskeleigenreflexe seitengleich mittellebhaft. Babinski beidseits negativ. Gangbild etwas unsicher, kleinschrittig.
Zusatzuntersuchungen
In der primär durchgeführten Computertomographie zeigten sich mehrere mediastinale Lymphadenopathien (scalenisch, oberes Mediastinum, infracarinär, peribronchial, zöliakal sowie retroperitoneal und mesenterial mit einem max. Durchmesser von 4,2 cm) (Abb. 1), eine ausgesprochene Splenomegalie (16 × 11,6 cm) mit nekrotischen Anteilen und schwerer Abgrenzbarkeit zum Magen, einem Befall der linken Nebenniere, intrapulmonalen Rundherden in allen Lungenlappen (max. Durchmesser 2,6 cm) sowie zwei intrahepatischen hypodensen Läsionen (Durchmesser 3,6 cm). Der Thymus war radiologisch unauffällig. In der Leberpunktion konnte die Diagnose eines diffusen grosszelligen B-Zell-Non-Hodgkin-Lymphoms (DLBCL) vom post-Keimzentrums-Typ (ABC-Typ) gestellt werden. Interessanterweise liess sich eine starke Expression sowohl für Bcl-2 als auch für das C-Myc-Protein nachweisen. Das Oberflächenprotein CD20 fiel positiv aus, neuroendokrine Marker waren negativ (Synaptophysin, Chromogranin, CD56, Pankreatin, TTF1, Napsin), der Proliferationsfaktor bei >90%. Laborchemisch war das LDH bei 1000 U/l (Norm: <225) und sank nach Immunchemotherapie auf 300–400 U/l. Elektrolyt- und Kreatininwertschwankungen im Verlauf normalisierten sich rasch ohne klinische Korrelation.
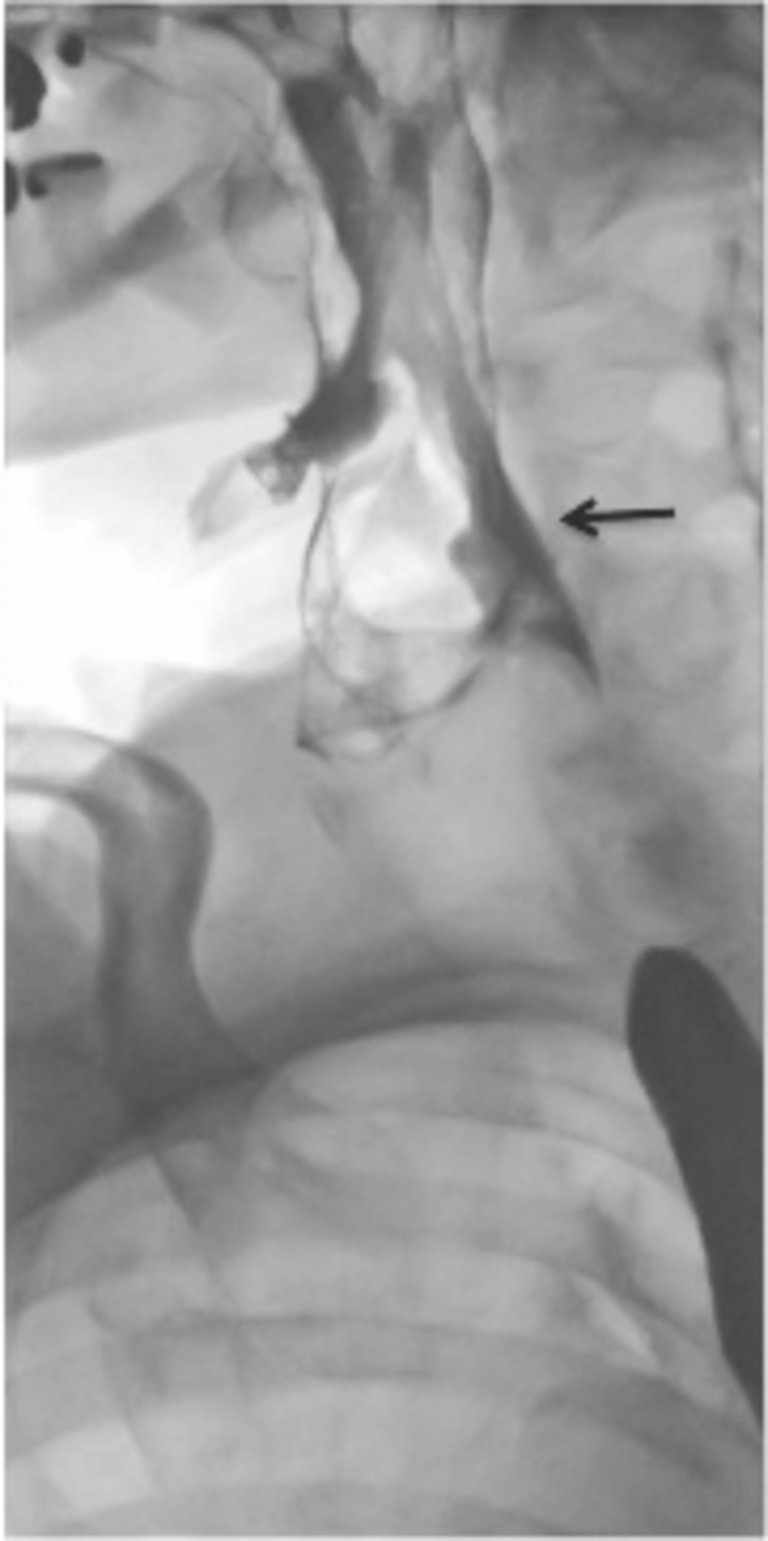
Abbildung 1: Computertomographie des Thorax: mediastinale Manifestation des Lymphomes. Pathologisch vergrösserte Lymphknoten in der oberen Thoraxapertur paratracheal mit einem maximalen Durchmesser bis 2,7 cm. Kleinere Lymphknoten peribronchial beidseits. Kein Nachweis eines Thymoms.
In der Magnetresonanztomographie des Gehirnes konnte eine Ischämie oder ein intrakranieller Lymphom-Befall ausgeschlossen werden. Eine Liquorpunktion verblieb unauffällig. In der repetitiven Stimulation des N. accessorius links mit 3 Hz zeigte sich kein pathologisches Amplitudendekrement. Die motorische Neurographie des N. medianus links ergab eine normwertige Amplitude, eine repetitive Stimulation mit 10 Hz sowie die Ableitung nach tonischer Innervation ergab kein Amplitudeninkrement. Im Breischluck (Ösophagus-Cinematographie) konnte radiologisch eine ausgeprägte Hypomobilität des Oro- und Hypopharynxes, jedoch mit regelrechten Kontraktionen des ösophagealen Sphinkters, dargestellt werden (Abb. 2).
Abbildung 2: Ösophagus-Cinématographie (Breischluck). Durch Hypomobilität verbleiben trotz mehrerer Nachschluckversuche Kontrastmittelreste sowohl auf dem Zungenrücken als auch im Oro-und Hypopharynx.
Therapie und Verlauf
Die Diagnosestellung des Lymphomes erfolgte kurz nach Spitaleintritt. Nach Diagnosestellung wurde ein erster Zyklus Chemotherapie mit Rituximab (700 mg) und Bendamustin (2 × 100 mg) nach einer Vorphase mit Vincristin (1 mg) und Solumedrol problemlos durchgeführt. Zu diesem Zeitpunkt hatte der Patient Essstörungen im Sinne von Schluckbeschwerden und zunehmender Aspiration. Diese wurden zunächst im Rahmen einer möglichen Lymphomkompression des N. recurrens interpretiert und eine Besserung nach Therapiestart erwartet. Bei jedoch zunehmender Unmöglichkeit einer peroralen Nahrungsaufnahme mit repetitivem Verschlucken und «drooling» durch die Nase wurde dem Patienten eine nasogastrale Sonde zur Kalorienzufuhr eingelegt.
Bei im weiteren Verlauf respiratorischer Verschlechterung mit zwischenzeitlichem peripherem Sättigungsabfall <90% konnten in der Verlaufscomputertomographie 3 Wochen nach Start der Chemotherapie eine Lungenembolie und Infiltrate ausgeschlossen werden. Es zeigte sich dabei eine «stable disease» nach den Kriterien für die Bewertung des Ansprechens der Behandlung bei soliden Tumoren (RECIST). Die bekannten Lymphadenopathien zeigten sich dabei überwiegend grössenregredient bis auf zwei leicht grössenprogrediente pulmonale Rundherde. Von den zwei Leberläsionen zeigte sich eine grössenregredient, die zweite grössenprogredient. Trotz nur minim erhöhten Entzündungszeichen und fehlendem Fieber begannen wir eine empirische Antibiotikatherapie, welche jedoch keine klinische Veränderung herbeiführen konnte. Bei weiterer Verschlechterung der Symptomatik mit akzentuierender Dysarthrie, Dysphagie und kompletter Immobilität war auf Wunsch des Patienten und der Angehörigen keine Therapie-Eskalation mehr gewünscht und wir stellten auf ein rein palliatives Procedere mit «best supportive care» um. Der Patient verstarb gut einen Monat nach initialer Hospitalisation. Auf eine Obduktion wurde auf Wunsch der Angehörigen des Patienten verzichtet.
Bei Verdacht auf eine paraneoplatische Ursache der bulbären Symptome in Form einer neuromuskulären Übertragungsstörung untersuchten wir Autoantikörper kurz vor Versterben des Patienten. Hierin zeigte sich ein diskret erhöhter Titer für Anti-Acetylcholin-Rezeptoren (AChR) (1,0 nmol/l, Norm: <0,5 nmol/l). Keine erhöhten Titer konnten für Anti-ZNS (Anti-Hu, Ri, Yo, Amphiphysin, CV2, Ta/Ma2, Ma1, SOX1, GAD65), Anti-Titin, Anti-Muskel-spezifische-Tyrosinkinase (MuSK), Anti-LGI1, Anti-CASPR2, Anti-«voltage-gated»-Calcium-Kanäle (VGCC) sowie Anti-«voltage-gated»-Kalium-Kanäle (VGKK) nachgewiesen werden.
Ursächlich für die unklare neurologische Symptomatik erhärtete sich somit der klinische Verdacht einer paraneoplastischen Myasthenia gravis. Wir vermuten daher, dass eine myasthene Symptomatik letztlich zum Tod des Patienten beigetragen hat.
Diskussion
Die neurologische Symptomatik mit Schwäche, Dysarthrie und Dysphagie trat erst im Verlauf nach Diagnosestellung des Lymphomes auf und exazerbierte trotz erfolgreicher Lymphomtherapie.
Als Ursache der Dysarhtrie und Dysphagie wurde initial ein raumfordernder Effekt durch die Lymphadenopathien mit etwaiger Rekurrensparese diskutiert. Diese Hypothesen befriedigten jedoch das klinische Bild, den Verlauf sowie die Befunde in den repetitiv durchgeführten Bildgebungen nicht. Ein Hinweis für eine paraneoplastische Myasthenia gravis lieferte der erhöhte Titer gegen AChR und die dazu passende Klinik. Die nur diskrete Erhöhung des AChR-Titers führten wir auf die erst nach verabreichter Steroid- und Chemo-/Immunotherapie durchgeführte Blutentnahme zurück, die unauffälligen elektrophysiologischen Untersuchungen schliessen eine Myasthenie nicht aus.
Myasthenia gravis als paraneoplastisches Syndrom eines Lymphomes
Myasthenia gravis ist eine autoimmunvermittelte Erkrankung, die durch Antikörper gegen den AChR an der neuromuskulären Endplatte der Skelettmuskulatur verursacht wird [1, 2]. 85% aller Patienten mit generalisierter Myasthenia gravis sind seropositiv für Acetylcholinrezeptor-Antikörper. Die Prävalenz der Myasthenia gravis beträgt 200–700 Fälle pro Million [2], die Inzidenz variiert, je nach Studie, zwischen 1,7–10,4 pro Million [1]. Bisher war die Myasthenia gravis vor allem als paraneoplastische Manifestation des Thymoms bekannt [2]. In einem Review von Rezania et al.[2] konnte jedoch eine Assoziation zwischen Myasthenia gravis und Lymphomen bzw. lymphoproliferativen Erkrankungen festgestellt werden. Genaue Untersuchungen bezüglich der Prävalenzen von Myasthenia gravis und Lymphomen wurden bisher aufgrund der Rarität noch nicht durchgeführt. Die häufigsten Assoziationen wurden mit Hodgkin-, T-Zell- oder Follikulären-Lymhomen beschrieben. Zeitpunkt der Diagnosestellung und Therapieverlauf verliefen hierbei sehr unterschiedlich [2].
Myasthenia gravis tritt häufiger auf bei Lymphomen mit einer mediastinalen Manifestation. Es wird vermutet, dass die räumliche Proximität zu den Thymozyten, die Proteine mit Ähnlichkeiten zum AChR exprimieren, einen Einfluss auf die Autoimmunreaktion haben. Die Entwicklung der Myasthenia gravis wird zudem durch gestörte Immunmechanismen begünstigt [2]. Durch eine Dysregulation der Lymphozytenselektion kommt es zu einer Depletion von regulatorischen T-Zellen und einer Vermehrung von autoreaktiven T-Zellen. In der Konsequenz kann eine ungehemmte Proliferation von selbstreaktiven B-Zellklonen stattfinden, welche spezifische Antikörper zum AChR produzieren [1–3].
Zur typischen Symptomatik der Myasthenia gravis gehören belastungsabhängige Muskelschwäche, Ptosis, Diplopie, Dysarthrie, Dysphagie, eine Schwäche der Gesichtsmuskulatur sowie eine Schwäche der Extremitäten- und der Stammmuskulatur. Meistens stellt sich innert 2 Jahren nach Symptombeginn eine generalisierte Schwäche ein [1]. Bei einer Mehrheit aller Patienten sind die okkulären Symptome mit Ptosis und Diplopie die ersten Krankheitszeichen [1, 2]. Im Zusammenhang mit Lymphomen sind in der Regel bulbäre Symptome wie Dysphagie, Dysarthrie und Kauschwierigkeiten sowie proximal beginnende Beinschwäche bei Krankheitsbeginn vorhanden [2], wie beim im Fallbericht beschriebenen Patienten.
Die myasthene Krise bezeichnet den Zustand, wenn der Patient aufgrund der generalisierten Schwäche der respiratorischen Muskulatur eine respiratorische Insuffizienz entwickelt [1].
Zur klinischen Diagnostik gehören der Edrophonium-Chlorid-Test (kurzwirksamer Acetylcholinesterase-Inhibitor) und der Ice-Pack-Test. Bei beiden wird sich im Falle einer Myasthenia gravis die Muskelschwäche für eine kurze Zeit verbessern. Elektrophysiologisch kann die repetitive Nervenstimulation getestet werden. Dabei kann man eine progressive Verminderung der Muskelsummenaktionspotential-Amplitude feststellen. Laborchemisch können die AChR-Antikörper im Serum bestimmt werden. Die Konzentrationen der AChR-Antikörper-Titer korrelieren hierbei nicht mit der Schwere der Symptome [1], führten jedoch in den bisher publizierten Fällen erst zur Diagnose [2]. Die nur diskrete AChR-Titererhöhung erklären wir uns durch die posttherapeutische Antikörperbestimmung. Es ist bekannt, dass eine Steroidtherapie, die Verabreichung des CD20-Antikörpers Rituximab sowie Chemotherapie zu Titerabnahmen führen [2]. Hierbei kann auch das fehlende Amplitudendekrement in der vierten Wochen nach Lymphomtherapie durchgeführten Nervenstimulation als therapiebedingt erklärt werden.
Die Therapie des Lymphoms bringt meistens auch eine Remission der Myasthenia gravis. Jedoch kann eine chemotherapeutische Behandlung einen Schub auslösen, indem sie einen Abfall von regulatorischen T-Zellen verursacht [2].
Im Rahmen der Akuttherapie bei Krankheitsexazerbation oder einer myasthenen Krise werden zur raschen Antikörperreduktion intravenös Immunglobuline verabreicht oder ein Plasmaaustausch vorgenommen [1, 2]. Zur Symptomkontrolle werden Cholinesteraseinhibitoren eingesetzt. Diese bringen aber weder eine komplette Symptomremission, noch nehmen sie Einfluss auf die Krankheitsprogression. Die Erhaltungstherapie besteht aus oralen Steroiden oder steroidsparenden Immunsuppressiva (Azathioprin, Myophenolat, Cylcosporin) [1, 2]. Oft kommt es unter der Therapie zu einer kompletten klinischen Remission der Myasthenia gravis, aber nicht zu einem kompletten Verschwinden der AChR-Antikörper. Die Prognose der Erkrankung ist abhängig von der Prognose des Lymphoms [2]. Der Verlauf des oben beschriebenen Patienten mit Akzentuierung der myasthenen Symptome trotz erfolgreicher Lymphombehandlung ist ungewöhnlich und bisher nicht beschrieben worden.
Das Wichtigste für die Praxis
• Leidet ein Patient mit Lymphom an unklaren bulbären Symptomen wie Dysarthrie und Dysphagie muss an ein paraneoplastisches Syndrom gedacht werden.
• Eine Chemotherapie kann bei paraneoplastischer Myasthenia gravis eine verstärkte myasthene Symptomatik bzw. Krise auslösen.
• Zur Reduktion der Latenz von Vorstellung bis Diagnosesicherung eines paraneoplastischen Syndromes mit Start einer adäquaten Therapie empfiehlt sich eine rasche multidisziplinäre Herangehensweise.
Verdankung
Wir danken Herrn Dr. med. Christopher Beynon (Leitender Arzt, Institut für Radiologie und Nuklearmedizin, Stadtspital Triemli, Zürich) für die Abbildungen.
Disclosure statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Correspondance
Dr. med. Andres Ort Stadtspital Triemli Birmensdorferstrasse 497 CH-8063 Zürich andresort[at]bluewin.ch
2 Rezania K, et al. Myasthenia gravis, an autoimmune manifestation of lymphoma and lymphoproliferative disorders: case reports and review of literature. Leuk Lymphoma. 2012;53(3):371–80.
3 Davis S, Schumacher MJ. Myasthenia gravis and lymphoma. A clinical and immunological association. JAMA. 1979;242(19):2096–7.