La cardiomyopathie hypertrophique et la cardiomyopathie dilatée sont des phénotypes myocardiques variables d’une multitude d’affections génétiques, non-génétiques et non-familiales. Un bilan étiologique et la prise en compte de certaines particularités thérapeutiques spécifiques revêtent une importance primordiale pour le traitement de ces patients.
Contexte
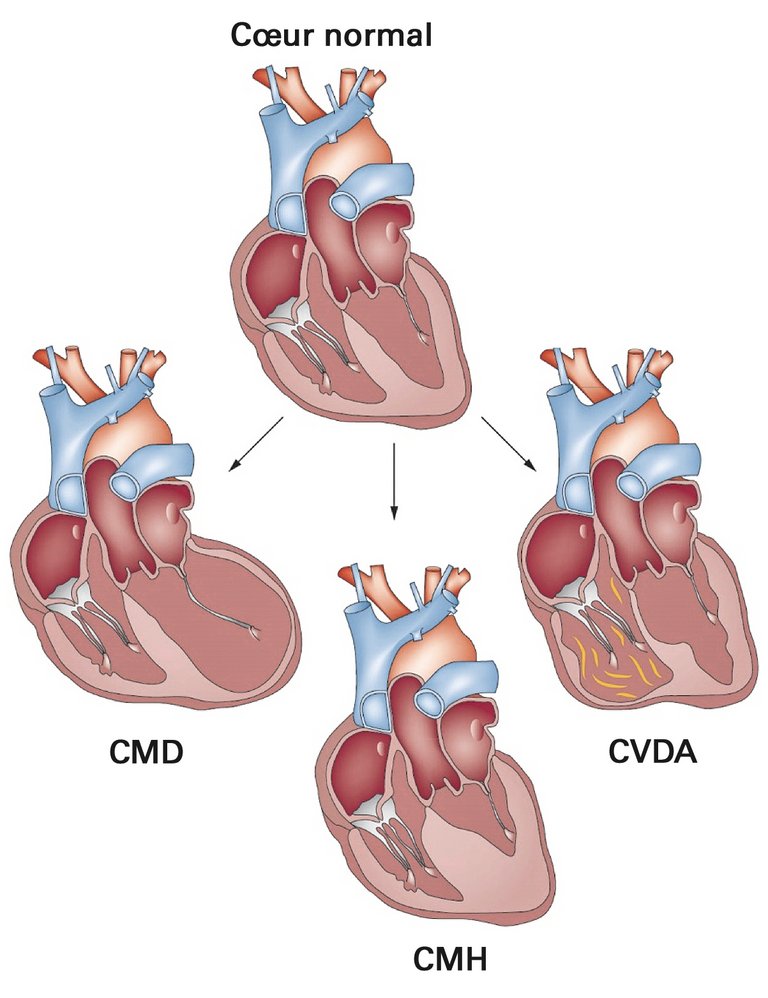
Le terme «cardiomyopathie» ne correspond pas à un diagnostic à proprement parler, mais bien plus à la description d’un phénotype myocardique morphologique et fonctionnel qui ne peut être justifié par une coronaropathie ou par des propriétés de remplissage altérées en raison d’une hypertension artérielle, d’une valvulopathie ou d’une cardiopathie congénitale. Sur la base des caractéristiques morphologiques et fonctionnelles, on distingue les cardiomyopathies hypertrophiques, dilatées, restrictives, ventriculaires droites arythmogènes ainsi que les cardiopathies non classées (fig. 1) [1].
Figure 1: Phénotypes morphologiques des cardiomyopathies les plus fréquentes. Alors que la cardiomyopathie dilatée (CMD) se définit par une augmentation du volume ventriculaire gauche ou biventriculaire et une baisse de la fonction systolique du ventricule gauche, la cardiomyopathie hypertrophique (CMH) se caractérise par une augmentation de l’épaisseur pariétale dans un ou plusieurs segments du ventricule gauche. La cardiomyopathie ventriculaire droite arythmogène (CVDA) est quant à elle caractérisée par un remplacement progressif du myocarde par du tissu adipeux et par une fibrose, le ventricule gauche étant parfois également touché. Adapted by permission from Macmillan Publishers Ltd: Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531–47, copyright 2013, https://www.nature.com/articles/nrcardio.2013.105 .
Dans le cadre des investigations diagnostiques, il est essentiel pour toutes les formes de délimiter les maladies familiales liées à des mutations (le plus souvent) monogéniques, responsables d’une pathologie myocardique primaire ou d’une affection systémique avec atteinte myocardique. En cas de susceptibilité correspondante, les cardiomyopathies non-familiales peuvent, quant à elles, être induites par divers facteurs déclenchants, souvent traitables (par ex. amyloses, virus, tachycardies) [1–3].
Afin de délimiter les formes familiales de cardiomyopathie, il est nécessaire de recueillir une anamnèse familiale chez tous les patients. Il convient tout particulièrement d’interroger le patient quant à la survenue de cas de mort subite cardiaque, d’insuffisance cardiaque, de nécessité de stimulateur cardiaque/défibrillateur automatique implantable (DAI) ou d’accident vasculaire cérébral chez des membres de sa famille à un âge jeune [2, 3]. La réalisation d’un arbre généalogique sur plusieurs générations permet, d’une part, de déceler une cause génétique de la cardiomyopathie et, d’autre part, d’identifier d’autres membres de la famille susceptibles d’être porteurs de la mutation. Etant donné que la majorité des cardiomyopathies sont transmises sur un mode autosomique dominant, la mutation est transmise aux descendants dans 50% des cas [4, 5]. Les cardiomyopathies transmises sur un mode autosomique récessif s’avèrent donc nettement plus rares et sont souvent l’expression d’une maladie systémique, comme par ex. une maladie lysosomale ou une ataxie de Friedreich [4].
Bien entendu, l’évaluation clinique d’une cardiomyopathie ne débute pas par l’identification d’un défaut génétique, mais par des symptômes spécifiques ou anomalies chez un patient. Il est souvent possible de déterminer l’étiologie au moyen de l’anamnèse, de l’examen clinique, d’examens de laboratoire ciblés et d’un examen d’imagerie cardiaque [2, 3]. L’imagerie cardiaque, et en particulier l’échocardiographie transthoracique, joue naturellement un rôle essentiel dans le diagnostic d’une cardiomyopathie. En présence d’indices évocateurs d’une cardiomyopathie, il convient en outre, dans le cadre de la procédure diagnostique de base, d’envisager la réalisation d’une imagerie par résonance magnétique (IRM) cardiaque afin d’évaluer la morphologie et la fonction cardiaques, l’ampleur de la fibrose et d’autres propriétés tissulaires [6].
Les sous-types phénotypiques les plus fréquents sont la cardiomyopathie hypertrophique (CMH) et la cardiomyopathie dilatée (CMD). Dans les lignes qui suivent, nous évoquerons les nouvelles connaissances relatives à l’étiologie de la CMH et de la CMD. En outre, pour les deux entités, nous présenterons également de nouvelles recommandations relatives au diagnostic et au traitement publiées récemment.
Cardiomyopathie hypertrophique
Clinique de la cardiomyopathie hypertrophique
La CMH n’est pas rare: sa prévalence s’élève à 0,02–0,2% et augmente avec l’âge [7, 8]. La CMH est définie comme un épaississement pathologique de la paroi du ventricule gauche ≥15 mm au niveau d’un ou plusieurs segments (indépendamment de la modalité d’imagerie), qui ne peut être entièrement expliqué par des propriétés de remplissage anormales [1, 4, 9]. Des difficultés diagnostiques peuvent émerger, notamment lorsqu’il s’agit de faire la distinction avec une hypertrophie myocardique suite à un entraînement physique intensif (cœur d’athlète [10]) ou en présence de maladies concomitantes, telles que l’hypertension artérielle ou une valvulopathie, car ces affections peuvent non seulement favoriser une hypertrophie ventriculaire gauche, mais aussi survenir indépendamment [4]. Pour les distinguer, il est souvent utile de recourir à des procédés d’imagerie spécialisés, comme par ex. une analyse détaillée de la fonction diastolique et une imagerie de déformation («strain imaging») à l’échocardiographie [11] ou un rehaussement tardif au gadolinium («late gadolinium enhancement») à l’IRM cardiaque [6]. Les patients d’âge avancé, atteints d’hypertension artérielle, présentent souvent une hypertrophie focale du septum basal (éperon septal) qui, par définition, n’est pas attribuable à la CMH bien que des symptômes similaires (dépendants de la charge) puissent survenir en raison d’obstacles à l’écoulement dans la chambre de chasse du ventricule gauche (CCVG) [4].
Chez les patients atteints de CMH, une mutation génétique en est responsable dans 60–70% des cas (tab. 1). Il s’agit majoritairement d’altérations génétiques à transmission autosomique dominante au niveau des protéines contractiles du sarcomère (fig. 2), telles que la chaîne lourde bêta de la myosine («beta-myosin heavy chain», MYH7) et la protéine C de liaison à la myosine («myosin-binding protein C», MYBPC3)[12, 13]. Dans la plupart des cas, des mutations génétiques sont à l’origine d’un phénotype avec hypertrophie septale asymétrique (90%) avec ou sans obstruction de la CCVG (appelée «cardiomyopathie obstructive hypertrophique», [CMOH]). Il convient d’en distinguer la CMH avec hypertrophie symétrique du ventricule gauche (5%), hypertrophie apicale (3%) et hypertrophie isolée de la paroi libre du ventricule gauche (2%).
Tableau 1: Principales étiologies de la cardiomyopathie hypertrophique (CMH).
Causes familiales
Anomalies génétiques fréquentes
Protéines contractiles du sarcomère
Anomalies génétiques rares
Maladies lysosomales (maladie de Fabry, maladie de Pompe)
Maladies mitochondriales
Syndromes (LEOPARD, Noonan, ataxie de Friedreich)
Amylose à transthyrétine mutée*
Hémochromatose
Anomalie génétique inconnue
Causes non génétiques
Amylose à transthyrétine sauvage ou amylose AL*
Obésité
Médicaments (corticostéroïdes, tacrolimus)
* La fonction de protéine de liaison amyloïde est assumée par la transthyrétine mutée dans le cas de l’amylose à transthyrétine mutée, par la transthyrétine non mutée dans le cas de l’amylose à transthyrétine sauvage et par les chaînes légères kappa ou lambda dans le cas de l’amylose AL.
Figure 2: Structure schématique d’un sarcomère. Le sarcomère forme la plus petite unité contractile des muscles et se compose pour l’essentiel de trois entités: le filament fin (majoritairement de l’actine), le filament épais (majoritairement de la myosine) et la protéine géante titine. Le filament fin est ancré dans la strie Z, alors que le filament épais est localisé au centre de la strie A. Au cours d’une activation, les têtes de myosine interagissent avec l’actine. La titine, qui s’étend de la strie Z à la strie M, a une action élastique dans la zone périphérique (strie I), comme un «ressort» moléculaire qui peut exercer une contre-force passive lors des stimuli de distension. Au niveau central (strie A), la titine n’est en revanche pas capable de se distendre car elle est en interaction étroite avec le filament épais, créant ainsi le centrage de la myosine avec le filament d’actine. Reproduced from Ottenheijm CA, van Hees HW, Heunks LM, Granzier H. Titin-based mechanosensing and signaling: role in diaphragm atrophy during unloading? Am J Physiol Lung Cell Mol Physiol. 2011;300(2):L161–6, by permission from the American Physiological Society. http://www.physiology.org/doi/10.1152/ajplung.00288.2010 .
Les symptômes de la CMH incluent la dyspnée, l’angor, l’intolérance à l’effort, les pré-syncopes et les syncopes (tab. 2). Dans le cadre de l’examen clinique, il est possible, en cas de CMOH, de retrouver à l’auscultation un souffle systolique d’éjection de basse fréquence au niveau de l’aorte, qui est renforcé en cas de réalisation d’une manœuvre visant à diminuer le remplissage ventriculaire gauche (par ex. passage de la position agenouillée à la position debout, manœuvre de Valsalva). En présence d’une insuffisance mitrale, un souffle systolique de régurgitation est en outre perçu au niveau de l’apex du cœur [4].
Tableau 2: Symptômes fréquents de la cardiomyopathie hypertrophique (CMH) et de la cardiomyopathie dilatée (CMD).
CMH
CMD
Dyspnée d’effort
Strictement dépendante de l’effort et de la fréquence
Symptôme principal, dépendante de l’effort
Orthopnée, dyspnée nocturne paroxystique
Rares, ou seulement à un stade avancé
Fréquentes, pathognomoniques
Angor
Fréquent
Occasionnel
Vertiges, pré-syncope, syncope
Typiques au cours de/après l’effort physique
Souvent d’origine orthostatique
Palpitations
Fréquentes
Occasionnelles
Diagnostic différentiel de la cardiomyopathie hypertrophique
Outre les mutations génétiques des protéines contractiles du sarcomère qui peuvent entraîner une hypertrophie myocardique, des maladies rares peuvent être responsables d’autres formes de CMH (env. 5% des patients avec CMH). En font partie des maladies de surcharge lysosomale (par ex. maladie de Fabry, maladie de Pompe), des maladies neuromusculaires (par ex. ataxie de Friedreich), des syndromes malformatifs (par ex. Noonan, LEOPARD) ainsi que des maladies mitochondriales, ces dernières étant principalement diagnostiquées chez les enfants en bas âge (tab. 1) [5, 14].
L’âge du patient au moment du diagnostic initial d’une cardiomyopathie est d’une importance capitale [2, 3]. Bien entendu, les maladies génétiques qui affectent par ex. le métabolisme ou la fonction mitochondriale, sont prépondérantes chez l’enfant. Par contre, la CMH provoquée par des mutations génétiques des protéines contractiles se manifestent le plus souvent uniquement au début de la puberté ou au début de l’âge adulte. Bien qu’une cause génétique ne soit pas exclue chez les patients de plus de 60 ans avec CMH nouvellement détectée, les étiologies non-familiales prédominent dans cette population (tab. 1). Un exemple typique en est la manifestation cardiaque d’une amylose à transthyrétine (amylose à TTR sauvage, autrefois appelée «amylose systémique sénile»), dans laquelle la transthyrétine non-mutée agit comme une protéine de liaison amyloïde [15]. Chez les patients de plus de 60 ans, l’amylose à TTR sauvage constitue un diagnostic différentiel majeur de la CMH [16], car elle est responsable principalement d’une atteinte cardiaque. Cependant, il convient de signaler que l’amylose à TTR familiale due à des mutations du gène de la transthyrétine est elle aussi fréquente chez les patients âgés avec CMH [16]. Env. 10% des patients âgés chez lesquels une CMH est diagnostiquée suite à une hypertrophie ventriculaire gauche souffrent en réalité d’une amylose cardiaque. Il est essentiel de faire la distinction entre ces deux étiologies, car le traitement n’est pas le même: ainsi, les antagonistes calciques, qui sont utiles en cas de CMH due à une hypertrophie myocardique, sont mal tolérés par les patients souffrant d’amylose à TTR. Il convient de suspecter une amylose cardiaque lorsque l’ECG ne révèle aucun signe d’hypertrophie ventriculaire gauche, voire lorsqu’un microvoltage du QRS («low QRS voltage») et un aspect de pseudo-infarctus sont présents. Outre les signes de l’insuffisance cardiaque, les manifestations cliniques d’une amylose sont les paresthésies et un syndrome du canal carpien (bilatéral), ainsi que typiquement des troubles orthostatiques [3, 4]. En plus de l’hypertrophie ventriculaire gauche, l’échocardiographie révèle une dilatation biatriale, des valves cardiaques épaissies, une hypertrophie du septum interauriculaire, un épanchement péricardique, une dysfonction diastolique de haut grade et, très typiquement, une diminution du «strain» longitudinal du myocarde. L’analyse échocardiographique de la déformation myocardique par «strain imaging» permet en outre de mettre en évidence un «apical sparing» caractéristique d’une atteinte ventriculaire gauche [17]. Les altérations typiques à l’IRM cardiaque sont quasiment diagnostiques. Le diagnostic est confirmé par mise en évidence immunohistochimique et par typage de l’amyloïde, le plus souvent au moyen d’une biopsie myocardique ou d’une biopsie du tissu adipeux sous-cutané.
Aspects essentiels relatifs au traitement de la cardiomyopathie hypertrophique
Les manifestations cliniques de la CMH sont particulièrement variables, pouvant aller du patient asymptomatique avec espérance de vie normale jusqu’au décès prématuré en raison d’arythmies ventriculaires, d’une insuffisance cardiaque terminale ou d’accidents vasculaires cérébraux. Une surveillance clinique minutieuse est capitale afin de détecter précocément les évolutions potentiellement dangereuses et initier un traitement adéquat (tab. 3) [18].
Tableau 3: Principes thérapeutiques de la cardiomyopathie hypertrophique (CMH).
Insuffisance cardiaque diastolique
Bêtabloquants
Antagonistes calciques (vérapamil, diltiazem)
Diurétiques (avec prudence)
Obstruction de la CCVG
Ablation septale, myectomie septale
Attention: diurétiques, inhibiteurs de l’ECA, nitrates, digoxine
Angor
Bêtabloquants
Antagonistes calciques (vérapamil, diltiazem)
Nitrates (avec prudence)
Tachyarythmies
Ventriculaires
DAI en prévention secondaire en cas de TV/FV documentée
DAI en prévention primaire en fonction de l’estimation du risque individuel (HCM Risk–SCD score)
Supraventriculaires
En cas de fibrillation auriculaire, anticoagulation orale avec antagoniste de la vitamine K (indépendamment du score CHA2DS2-VASc)
Contrôle de la fréquence (bêtabloquants, vérapamil, diltiazem)
CCVG = chambre de chasse du ventricule gauche; DAI = défibrillateur automatique implantable; TV = tachycardie ventriculaire; FV = fibrillation ventriculaire.
Insuffisance cardiaque
Les patients atteints de CMH peuvent développer des symptômes d’insuffisance cardiaque, car le remplissage diastolique du ventricule gauche est altéré en raison de la masse musculaire accrue, ce qui entraîne une augmentation de la pression de remplissage télédiastolique. Le patient présente alors les manifestations typiques d’une insuffisance cardiaque à fraction d’éjection préservée (ICFEP). L’ICFEP est définie comme une insuffisance cardiaque cliniquement manifeste se caractérisant par une fraction d’éjection ventriculaire gauche (FEVG) ≥50% et présence concomitante d’une dysfonction diastolique ou par une cardiopathie structurelle pertinente, ainsi que par une élévation des peptides natriurétiques [19]. Des substances à action bradycardisante et lusitrope positive (c.-à-d. qui améliorent la relaxation cardiaque) sont utilisées pour améliorer la fonction diastolique. En font partie les bêtabloquants et les antagonistes calciques (vérapamil ou diltiazem) [4]. Une surveillance clinique est toutefois recommandée, car le volume d’éjection systolique du ventricule gauche n’est sujet qu’à de faibles variations, l’augmentation du débit cardiaque dépendant ainsi principalement de la fréquence cardiaque. Pour des considérations similaires, les diurétiques devraient eux aussi être utilisés avec prudence. L’administration de digoxine n’est d’une manière générale pas recommandée [20]. Les médicaments vasodilatateurs, comme par ex. les inhibiteurs de l’enzyme de conversion de l’angiotensine (ECA), devraient également être administrés avec prudence car, en raison de leur effet de diminution de la post-charge, ils réduisent la tension pariétale du ventricule gauche et ainsi, son volume. Cela explique qu’en cas d’obstruction de la CCVG, il se produit une augmentation de la pression ventriculaire gauche et donc des gradients dans la CCVG afin de maintenir le volume d’éjection systolique. La même détérioration physiopathologique peut arriver suite à la consommation d’alcool, en raison de l’action vasodilatatrice de cette substance. Chez les patients atteints de CMH qui se présentent avec un œdème pulmonaire et une hypertension artérielle, il convient d’utiliser des bêtabloquants et des vasoconstricteurs, car les vasodilatateurs peuvent s’avérer fatals dans cette situation [4].
Obstruction de la CCVG: cardiomyopathie obstructive hypertrophique
L’hypertrophie du septum interventriculaire basal a la particularité d’entraîner une obstruction de la CCVG chez env. un tiers des patients atteints de CMH. Elle est détectée par échocardiographie doppler sous forme de gradient dynamique; elle peut être responsable de syncopes, d’une insuffisance cardiaque et d’une intolérance à l’effort, et elle augmente également le risque de mort subite cardiaque. En outre, le feuillet mitral antérieur peut venir au contact du septum durant la systole (phénomène appelé «mouvement systolique antérieur» ou «systolic anterior movement» [SAM]) par déplacement antérieur des muscles papillaires et effet d’aspiration lié au flux (effet Venturi) dans la CCVG. Il en résulte un trouble de la coaptation de la valve mitrale, avec insuffisance mitrale consécutive qui renforce la dyspnée. Un gradient (provoqué) ≥50 mm Hg dans la CCVG est généralement considéré comme pertinent sur le plan hémodynamique [4].
Une myectomie septale visant à traiter une obstruction pertinente de la CCVG devrait être envisagée si les symptômes cliniques persistent malgré le traitement médicamenteux pour l’insuffisance cardiaque. Une approche interventionnelle est possible, basée sur l’ablation du septum par injection d’alcool dans la première branche septale [21]. En particulier lorsque des altérations morphologiques au niveau de la valve mitrale requièrent une reconstruction adéquate, l’obstruction peut également être traitée par voie chirurgicale [22]. La complication la plus fréquente pour les deux approches est l’induction d’un bloc auriculo-ventriculaire, qui nécessite l’implantation d’un stimulateur cardiaque. Les résultats de ces deux méthodes de traitement sont comparables, le taux de complications (mortalité, nécessité d’implantation d’un stimulateur cardiaque, hémorragies, insuffisance rénale) dépendant principalement de l’expérience de l’opérateur [23, 24].
Angor
Les ischémies myocardiques consécutives à une réserve coronaire réduite, à dysfonction microvasculaire ou à obstruction de la CCVG représente un problème fréquent chez les patients souffrant de CMH. En fonction de la probabilité clinique pré-test, il convient, en cas d’angor, d’exclure, par voie invasive ou par tomodensitométrie (TDM) coronaire, une coronaropathie causale. Les bêtabloquants et les antagonistes calciques peuvent être utilisés pour réduire les besoins myocardiques en oxygène et ainsi réduire l’ischémie [4]. Les nitrates peuvent être prescrits chez les patients sans gradient dans la CCVG; par contre, ils doivent uniquement être administrés sous surveillance clinique attentive en cas de CMOH.
Tachyarythmies ventriculaires
Chez les patients atteints de CMH, la mortalité cardiovasculaire s’élève à 1–2% par an et est attribuable à une insuffisance cardiaque, à des thromboembolies ou à une fibrillation ventriculaire spontanée. L’évaluation du risque d’arythmies ventriculaires malignes est dès lors essentielle. Le score HCM Risk-SCD ([«sudden cardiac death»]) a été développé à cet effet. Le score de risque repose sur l’examen clinique, sur l’anamnèse familiale, sur l’imagerie cardiaque et sur l’ECG de longue durée; il estime le risque individuel à 5 ans de mort subite cardiaque [25]. Il est possible d’utiliser ce score comme base pour évaluer l’implantation d’un DAI en prévention primaire, tout en tenant compte de l’âge, des comorbidités et des facteurs psychologiques. Il convient de mentionner que le score n’a à ce jour pas été validé de manière prospective. De même, l’utilisation en prévention primaire d’anti-arythmiques n’a guère été étudiée. Etant donné que les arythmies sont déclenchées par des stimuli adrénergiques, il est recommandé aux patients atteints de CMH de ne pratiquer aucun sport de compétition [4]. Chez les patients ayant survécu à une arythmie ventriculaire, l’implantation d’un DAI en prévention secondaire est indiquée dans la plupart des cas [4].
Fibrillation auriculaire
Les arythmies supraventriculaires, et en particulier la fibrillation auriculaire, sont plus fréquentes que les arythmies ventriculaires chez les patients atteints de CMH. L’incidence annuelle de la fibrillation auriculaire et des thromboembolies s’élève à env. 25%, ces évènements étant associés à une évolution défavorable [26]. La survenue d’une fibrillation auriculaire est corrélée avec la taille de l’oreillette gauche [27], raison pour laquelle un dépistage tous les 6-12 mois par ECG de longue durée est recommandé à partir d’un diamètre de 45 mm. En raison du risque élevé de thromboembolie associé à la fibrillation auriculaire en cas de CMH, il est généralement recommandé d’utiliser de manière illimitée dans le temps des antagonistes de la vitamine K (ou des anticoagulants oraux non antivitamine K) et ce, indépendamment du score CHA2DS2-VASc [4,26].
Suivi
Chez les patients cliniquement stables atteints de CMH, un suivi cardiologique par ECG, échocardiographie et ECG de longue durée est recommandé tous les 1–2 ans. En cas de changements des symptômes, le contrôle doit être avancé en conséquence. Chez les apparentés au premier degré d’un patient atteint de CMH sans mise en évidence d’une mutation génétique, il est recommandé de rechercher une affection par ECG et échocardiographie. Etant donné qu’un développement ultérieur de la maladie ne peut être exclu en dépit d’un examen cardiologique normal en raison d’une pénétrance dépendante de l’âge, des contrôles réguliers doivent être assurés (en fonction de l’âge et de la sévérité de la CMH chez le patient index). D’une manière générale, un intervalle de 2–5 ans est recommandé, pour autant qu’aucun symptôme spécifique à la maladie n’apparaisse entre-temps.
Cardiomyopathie dilatée
Un phénotype myocardique variable
La prévalence de la CMD s’élève à env. 0,04% et est ainsi comparable à celle de la CMH [7, 28]. Elle se définit d’une part par une augmentation du volume télédiastolique ventriculaire gauche (ou biventriculaire; normalisé pour la surface corporelle et le sexe) et par une réduction de la FEVG d’autre part [1]. Avant la pose définitive du diagnostic, les anomalies devraient être confirmées par une modalité d’imagerie alternative ou dans le cadre d’un examen de suivi [1]. La CMD est le plus souvent diagnostiquée au cours de la 4e ou 5e décennie de vie. Les principaux symptômes incluent la dyspnée d’effort, l’orthopnée et la dyspnée paroxystique nocturne (tab. 2). Le spectre clinique et l’évolution naturelle de la CMD sont vastes: cela s’explique d’une part par la grande hétérogénéité des étiologies génétiques et non-familiales (tab. 4). D’autre part, la maladie évolue généralement de manière silencieuse dans un premier temps, par ex. chez les porteurs d’une mutation génétique, avec une pénétrance et une expression de la maladie très variables au sein des familles [5, 14, 29]. Avec l’expression croissante du phénotype, les patients peuvent présenter initialement soit une dilatation isolée du ventricule gauche avec FEVG préservée soit, à l’inverse, une diminution de la FEVG avec dimensions ventriculaires encore normales («cardiomyopathie hypokinétique non dilatée») [29]. C’est uniquement en cas de progression supplémentaire de ce phénotype intermédiaire que le tableau de CMD à proprement parler se manifestera.
Tableau 4: Principales étiologies de la cardiomyopathie dilatée (CMD).
Causes familiales
Anomalies génétiques fréquentes
Protéines structurales du sarcomère (titine, lamine)
Anomalies génétiques rares
Autres protéines du sarcomère (myosine, troponine T, protéine de liaison à la myosine, RBM20, myopalladine, pompe à potassium, BAG3, phospholamban)
Affections neuromusculaires (dystrophie musculaire de Duchenne, dystrophie musculaire de Becker, dystrophie myotonique)
* Pour les causes non génétiques, il s’agit souvent de facteurs déclenchants qui, en cas de prédisposition génétique correspondante, entraînent une expression de la CMD.
Diagnostic différentiel de la cardiomyopathie dilatée
La CMD est un phénotype commun à de nombreuses affections non-familiales et familiales (génétiques) [5]. Dans le cadre du diagnostic différentiel d’une CMD, le contexte clinique fournit des indications essentielles, les moyens diagnostiques à employer (analyses de laboratoire, imagerie) devant être déterminés individuellement en fonction de chaque patient [15, 29]. Les principales causes de CMD sont résumées dans le tableau 4. Il s’agit souvent d’une myocardite, d’une inflammation chronique en raison d’une infection virale persistante ou d’une réponse auto-immune induite par un virus. L’IRM cardiaque s’est imposée comme examen diagnostique majeur pour la CMD causée par une myocardite. En revanche, la nécessité de réaliser une biopsie endomyocardique en vue d’un examen histologique/immunohistochimique et de la mise en évidence d’un virus est sujette à controverse [15, 30, 31]. L’IRM, quant à elle, peut également fournir des indications quant à la présence d’une sarcoïdose ou d’une cardiomyopathie ventriculaire droite arythmogène (CVDA) avec atteinte ventriculaire gauche [6]. Chez les patients avec réduction de la FEVG nouvellement diagnostiquée, il convient de réaliser une angiographie coronaire afin d’exclure une coronaropathie causale, car env. 5% des patients présentent une coronaropathie sévère et tirent d’énormes bénéfices d’une revascularisation. En cas d’évaluation invasive, un cathétérisme cardiaque droit doit être réalisé simultanément, car cet examen fournit des informations supplémentaires précieuses pour orienter le traitement (par ex. intensification de la diminution de la précharge en cas de pressions de remplissage élevées).
A la différence de la CMH, pour laquelle les formes familiales prédominent, une cause génétique a pendant longtemps uniquement été soupçonnée dans env. 20% des cas de CMD [5,15]. La compréhension toujours plus fine des causes génétiques des cardiomyopathies est attribuable aux progrès technologiques accomplis au niveau des méthodes de séquençage génétique. Aujourd’hui, 30–50% des cas de CMD sont attribués à une cause génétique [15]. A cet égard, il est élémentaire que le séquençage de très grands gènes tels que la titine, la plus grande protéine dans le corps humain, est uniquement possible depuis quelques années [32]. En tant que protéine structurale, la titine forme la «colonne vertébrale» des sarcomères (fig. 2) [33, 34]. Elle remplit trois tâches essentielles: (1.) la titine forme une armature qui centre les têtes de myosine entre les filaments d’actine et garantit ainsi une transmission de force optimale; (2.) la titine forme le «ressort» qui assure un retour passif de l’appareil contractile après la contraction; (3.) la phosphorylation de la titine a une influence déterminante sur la rigidité et donc la fonction diastolique du myocarde [32]. Il est dès lors aisément compréhensible que les altérations au niveau de cette protéine structurale puissent être à l’origine d’une perturbation de la fonction de pompe et d’une dilatation du ventricule gauche. Les mutations du gène de la titine sont les causes familiales les plus fréquentes de la CMD (jusqu’à 25% des patients) [33,35]. En raison de la taille du gène, son séquençage est techniquement complexe, raison pour laquelle une analyse ne peut actuellement pas encore être réalisée de manière routinière. S’y ajoute la difficulté suivante: chez les patients avec CMD d’origine génétique, il y a souvent plus d’une mutation potentiellement causale qui peut être mise en évidence [35]. Ceci pourrait expliquer l’expression souvent très variable de la maladie au sein d’une même famille, mais cela complique en conséquence l’interprétation des tests génétiques, qui sont actuellement peu utiles pour les patients individuels.
Grâce à la compréhension croissante des causes génétiques, il est aussi désormais connu qu’en présence de facteurs déclenchants correspondants, les mutations génétiques déterminent la susceptibilité individuelle vis-à-vis de la manifestation phénotypique d’une CMD. Des mutations du gène de la titine ont par ex. pu être identifiées comme un facteur prédisposant au développement d’une cardiomyopathie du péripartum [36]. Il pourrait également en être de même pour la CMD en cas de consommation importante d’alcool. Etant donné que seule une minorité des patients avec une consommation accrue d’alcool développent une CMD et qu’il n’y a pas de corrélation entre la quantité d’alcool consommée et l’atteinte myocardique, le développement d’une cardiomyopathie doit être déterminé par une susceptibilité individuelle (d’origine génétique) [37].
Aspects essentiels relatifs au traitement de la cardiomyopathie dilatée
Dans les formes non-familiales de CMD, il est essentiel de traiter dans la mesure du possible le facteur déclenchant (abstinence totale de substances nocives, réévaluation du bénéfice de la poursuite d’une chimiothérapie, etc.) ou la maladie sous-jacente (maladie endocrinienne, collagénose, etc.), ce qui peut s’avérer particulièrement complexe dans certains cas. En outre, des approches thérapeutiques cardiaques spécifiques devraient être mises en œuvre chez tous les patients.
Insuffisance cardiaque
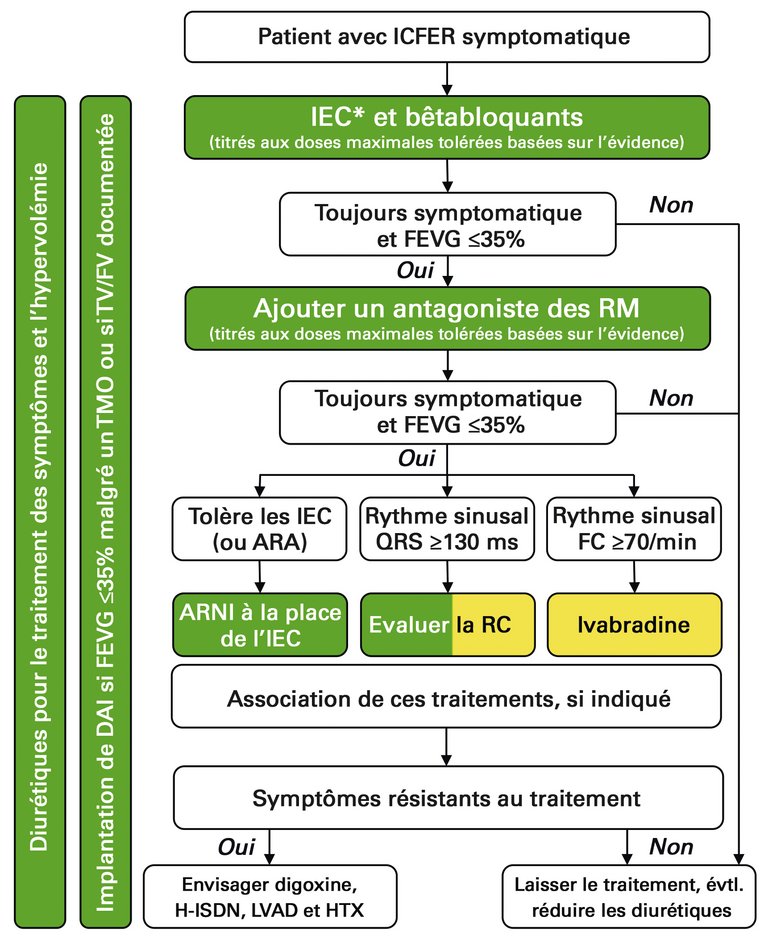
Les patients atteints de CMD souffrent par nature d’insuffisance cardiaque («insuffisance cardiaque à fraction d’éjection réduite», ICFER, définie comme une FEVG <40%) et requièrent un traitement optimal correspondant (fig. 3) [19]. Les recommandations thérapeutiques présentées, issues de la «European Society of Cardiology» reposent sur le traitement connu impliquant une intensification progressive, mais elles contiennent quelques nouveautés notables. La base du traitement réside dans l’association d’un inhibiteur de l’ECA et d’un bêtabloquant, dont la posologie devrait être augmentée progressivement jusqu’à ce que les doses maximales tolérées basées sur l’évidence soient atteintes. Les sartans ne constituent pas une alternative équivalente aux inhibiteurs de l’ECA, mais ils peuvent et doivent être utilisés en cas d’intolérance aux inhibiteurs de l’ECA. Tandis que ces médicaments exercent une influence positive sur la morbidité et la mortalité, les diurétiques devraient être utilisés comme traitement symptomatique et leur posologie devrait être augmentée jusqu’à la dose permettant de maintenir l’euvolémie, moyennant une réévaluation clinique régulière. Si le patient reste symptomatique (c.-à-d. classe fonctionnelle NYHA II–IV) et si la FEVG est ≤35%, il est recommandé de procéder à une intensification thérapeutique avec un antagoniste des récepteurs minéralocorticoïdes afin de réduire la mortalité et le taux d’hospitalisations. La spironolactone et l’éplérénone n’ont ainsi toujours pas leur place dans le traitement initial de l’insuffisance cardiaque. Chez les patients suivant déjà ce traitement intensifié, l’utilisation d’un inhibiteur du récepteur de l’angiotensine et de la néprilysine (ARNI) représente depuis peu une nouvelle option thérapeutique complémentaire. La première substance disponible de cette classe, LCZ696 (Entresto®), est une association fixe de valsartan et sacubitril. L’inhibition de la néprilysine par le sacubitril empêche la dégradation des peptides natriurétiques (ANP, BNP), favorisant ainsi la diurèse, la natriurèse et la relaxation myocardique [38]. L’étude PARADIGM-HF est parvenue à démontrer un effet positif de LCZ696 sur le taux d’hospitalisations et la mortalité par rapport au traitement standard par énalapril [39]. Environ un tiers des patients ne tolèrent pas LCZ696 en raison de l’hypotension induite en conséquence. Dès lors, l’utilisation de LCZ696 est uniquement recommandée chez les patients avec une pression artérielle systolique ≥100 mm Hg. En raison de l’effet diurétique du sacubitril, une réduction des diurétiques est souvent nécessaire. L’association d’un ARNI et d’un inhibiteur de l’ECA est contre-indiquée en raison d’un risque d’angiœdème; l’inhibiteur de l’ECA doit être interrompu 36 heures avant l’initiation de LCZ696. Les valeurs de BNP augmentent sous traitement par inhibiteurs de la néprilysine du fait du mécanisme d’action de ces substances, raison pour laquelle seules les valeurs de NT-proBNP peuvent être utilisées pour évaluer l’évolution de l’insuffisance cardiaque.
Figure 3: Algorithme thérapeutique pour un patient présentant une insuffisance cardiaque symptomatique (classe NYHA II-IV) à fraction d’éjection ventriculaire gauche réduite (ICFER, FEVG <40%). En vert se trouvent les recommandations de classe I et en jaune les recommandations de classe IIa selon les directives actuelles de l’«European Society of Cardiology». * Utiliser des ARA au cas où les inhibiteurs de l’ECA ne sont pas tolérés ou sont contre-indiqués; IEC = inhibiteurs de l’enzyme de conversion de l’angiotensine; ARA = antagonistes des récepteurs de l’angiotensine; ARNI = inhibiteurs du récepteur de l’angiotensine et de la néprilysine; RC = resynchronisation cardiaque; H-ISDN = hydralazine et dinitrate d’isosorbide; FC = fréquence cardiaque; HTX = transplantation cardiaque; DAI = défibrillateur automatique implantable; LVAD = dispositif d’assistance ventriculaire gauche; FEVG = fraction d’éjection ventriculaire gauche; RM = récepteur des minéralocorticoïdes; TMO = traitement médicamenteux optimal; FV = fibrillation ventriculaire; TV = tachycardie ventriculaire. From Figure 7.1. from Ponikowski P, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2016; 37(27):2129–00. doi: 10.1093/eurheartj/ehw128 . Published by Oxford University Press on behalf of the European Society of Cardiology. All rights reserved. Available online at: https://academic.oup.com/eurheartj/article/37/27/2129/1748921 . This figure is not covered by the Open-Access licence of this publication. For permissions contact Journals.permissions@OUP.com. Translated and reprinted with permission of Oxford University Press on behalf of the European Society of Cardiology. OUP and the ESC are not responsible or in any way liable for the accuracy of the translation. MR Meyer and FR Eberli are solely responsible for the translation in this publication. Please visit: www.escardio.org/Guidelines/Clinical-Practice-Guidelines/Acute-and-Chronic-Heart-Failure .
Un traitement de resynchronisation cardiaque doit être envisagé en cas de complexe QRS élargi et le contrôle de la fréquence cardiaque par ivabradine doit être considéré en cas de fréquence cardiaque >70/min malgré un traitement de base optimal. Enfin, un traitement par digoxine ou dinitrate d’isosorbide-hydralazine, ainsi que par dispositif d’assistance ventriculaire gauche ou transplantation cardiaque, peut être évalué aux stades terminaux (fig. 3). Chez les patients atteints de CMD, le suivi doit être déterminé selon le cas et dépend tout particulièrement de l’évolution de la maladie, de la nécessité d’adaptation thérapeutique et de la compréhension du patient par rapport à sa propre maladie.
Tachyarythmies et cardiomyopathie dilatée
Les options disponibles pour le traitement de l’insuffisance cardiaque chez les patients atteints de CMD (inhibiteurs de l’ECA, bêtabloquants, antagonistes des récepteurs minéralocorticoïdes, sacubitril/valsartan et traitement de resynchronisation cardiaque) réduisent considérablement le risque de mort cardiaque subite [19]. En cas de FEVG ≤35% et ce, malgré la prise d’un tel traitement, les recommandations thérapeutiques internationales préconisent en outre l’implantation d’un DAI en prévention primaire (fig. 3) [19]. Ces recommandations reposent en grande partie sur des études, qui ont principalement inclus des patients présentant une fraction d’éjection réduite de par une coronaropathie. L’intérêt du DAI chez les patients atteints de CMD non-ischémique sous traitement médicamenteux optimal de l’insuffisance cardiaque a été remis en question suite à la publication récente de l’étude DANISH. En particulier chez les patients de plus de 68 ans avec comorbidités supplémentaires, le DAI en prévention primaire ne semble pas profitable en termes de mortalité cardiovasculaire et de mortalité globale [40]. En revanche, chez les patients avec CMD, qui ont survécu à une arythmie ventriculaire pertinente sur le plan hémodynamique, l’implantation d’un DAI en prévention secondaire est recommandée, pour autant qu’ils aient une espérance de vie >1 an [19].
Les arythmies supraventriculaires, et en particulier la fibrillation auriculaire, constituent un problème fréquent chez les patients atteints de CMD. Le traitement, et avant tout l’anticoagulation, doit être effectué conformément aux recommandations actuelles [41]. L’évaluation du risque de thromboembolie revêt une importance capitale; l’utilisation d’anticoagulants oraux non antivitamine K (à privilégier par rapport aux antagonistes de la vitamine K) doit se baser sur le score CHA2DS2-VASc chez les patients atteints de CMD. Par contre, il n’est pas recommandé d’initier une anticoagulation sur la seule base d’une mauvaise FEVG.
Cardiomyopathies familiales: rôle des tests génétiques
En l’état actuel des connaissances, la mise en évidence d’une mutation responsable par la génétique moléculaire ne se trouve pas au premier plan pour la confirmation du diagnostic et la prise en charge clinique d’une CMH ou d’une CMD, qui ne sont pas l’expression d’une maladie systémique rare [4,42]. L’intérêt des tests génétiques réside bien plus dans le dépistage et la détection précoce (préclinique) de la maladie chez des apparentés directs de patients dont le diagnostic est certain, dès lors que ceux-ci sont disposés à réaliser un test génétique [4, 42, 43]. Si une mutation causale parvient à être mise en évidence chez le patient index, le test génétique des apparentés directs constitue une approche (économiquement) efficace, car des examens cardiologiques répétés ne seront pas nécessaires en cas de test négatif chez les apparentés [44]. Il convient toutefois de nuancer ce propos en mentionnant que la mise en évidence d’une mutation génétique via l’analyse pré-spécifiée d’un panel de variants connus (par ex. gènes des protéines du sarcomère) n’aboutit pas toujours, car la maladie peut également être provoquée par une anomalie génétique encore inconnue (et donc non comprise dans le test). Une consultation génétique, portant également sur les conséquences psychologiques, sociales, éthiques et légales, d’un test génétique est dans tous les cas indispensable [4].
Il n’est pas étonnant que les mutations génétiques dans les protéines du sarcomère puissent parfois conduire à des phénotypes myocardiques variables. Par exemple, des mutations dans les acides aminés avoisinants du gène MYH7 peuvent entraîner une CMH ou une CMD [45, 46]. D’un autre côté, des mutations du gène de la titine, principale cause génétique de CMD, peuvent également être mises en évidence chez env. 1% des patients atteints de CMH [33]. La compréhension croissante des principes de génétique moléculaire remet donc en question la classification traditionnelle des cardiomyopathies en fonction de leurs caractéristiques morphologiques et fonctionnelles, bien que ces catégories aient fait leurs preuves dans la pratique clinique quotidienne. A l’inverse, la connaissance de l’hétérogénéité génétique des cardiomyopathies familiales, avec de nombreux gènes potentiellement impliqués pouvant eux-mêmes présenter des mutations souvent rares et très diverses, revêt une importance centrale lors de l’utilisation des tests génétiques [5, 14]. Ainsi, >1400 variants génétiques ont déjà été associés à la CMH. Cela illustre le niveau de difficulté de la prise de décision clinique sur la base de la mise en évidence de mutations pathogènes, de variations bénignes et d’altérations génétiques à la signification indéterminée [42].
Perspectives
En décalage par rapport avec la fréquence des cardiomyopathies, et en particulier de la CMH, la compréhension actuelle de la pathogenèse et le traitement de ces affections reposent essentiellement sur des études observationnelles et des études de cohorte avec des effectifs de patients relativement faibles [47]. Il sera dès lors essentiel de disposer à l’avenir d’études cliniques randomisées prospectives. L’exploration plus poussée des principes génétiques des cardiomyopathies devrait également aboutir à une meilleure compréhension de la physiopathologie, des distinctions pronostiques et donc des implications thérapeutiques futures. Il a ainsi pu être montré que les variations dans le gène de la titine constituaient un facteur prédisposant chez les femmes atteintes de cardiomyopathie du péripartum [36]. Il semble également en être de même chez les patients atteints de CMH: en présence de facteurs de risque supplémentaires (par ex. hypertension artérielle, obésité, diabète sucré, syndrome d’apnée obstructive du sommeil), des mutations génétiques spécifiques peuvent prédisposer à une expression phénotypique de l’hypertrophie myocardique [48]. Chez les patients avec un génotype correspondant, il convient peut-être de traiter en premier lieu le facteur déclenchant, comme par ex. l’obtention d’un contrôle strict et précoce de la pression artérielle. La compréhension croissante des fondements génétiques pourrait aussi permettre à l’avenir de développer des traitements génétiques (par ex. «antisense oligonucleotides, gene editing») ou des traitements spécifiques aux différents patients [49].
L’essentiel pour la pratique
• La cardiomyopathie hypertrophique (CMH) et la cardiomyopathie dilatée (CMD) sont des phénotypes myocardiques qui ne sont pas la conséquence d’une coronaropathie avancée, d’une hypertension artérielle, de valvulopathies ou d’une cardiopathie congénitale.
• La CMH est souvent provoquée par une mutation des protéines contractiles du sarcomère. La CMD est un phénotype commun à une multitude de maladies non-familiales et de maladies génétiques, les mutations dans le gène de la titine (une protéine structurale du sarcomère) ayant récemment été identifiées comme principale cause familiale.
• L’étiologie spécifique de la cardiomyopathie est identifiable grâce au contexte clinique (anamnèse, arbre généalogique), à des analyses de laboratoire ciblées et à l’imagerie cardiaque (échocardiographie, IRM cardiaque).
• Les approches thérapeutiques spécifiques de la CMH comprennent le traitement de l’insuffisance cardiaque, de l’angor, de l’obstruction de la chambre de chasse du ventricule gauche (CCVG) et des tachyarythmies, y compris l’évaluation d’un traitement par défibrillateur automatique implantable (DAI). Les médicaments de choix sont les bêtabloquants et les antagonistes calciques; les vasodilatateurs ne doivent quant à eux pas être utilisés en cas d’obstruction pertinente de la CCVG.
• Outre l’élimination d’un éventuel facteur déclenchant ou le traitement de la maladie sous-jacente, le traitement de la CMD comprend toujours une prise en charge optimale de l’insuffisance cardiaque.
• En cas de cardiomyopathie, les analyses génétiques sont utiles lorsqu’elles recherchent de façon ciblée une mutation sur la base d’un diagnostic clinique de suspicion ou lorsqu’elles sont utilisées pour le dépistage des apparentés directs de patients présentant une mutation pathogène avérée.
Disclosure statement
Les auteurs n’ont pas déclaré d’obligations financières ou personnelles en rapport avec l’article soumis.
Correspondance
Dr méd. Matthias Meyer Klinik für Kardiologie Stadtspital Triemli Birmensdorferstrasse 497 CH-8063 Zürich matthias.meyer[at]triemli.zuerich.ch
Références
1 Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270–6.
2 Rapezzi C, Arbustini E, Caforio AL, Charron P, Gimeno-Blanes J, Helio T, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34(19):1448–58.
3 Lopes LR, Elliott PM. New approaches to the clinical diagnosis of inherited heart muscle disease. Heart. 2013;99(19):1451–61.
4 Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733–79.
5 Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011;364(17):1643–56.
6 Tummala LS, Young RK, Singh T, Jani S, Srichai MB. Role of non-invasive imaging in the work-up of cardiomyopathies. Curr Atheroscler Rep. 2015;17(3):486.
7 Codd MB, Sugrue DD, Gersh BJ, Melton LJ, 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–84. Circulation. 1989;80(3):564–72.
8 Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med. 1998;339(6):364–9.
9 Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124(24):2761–96.
10 Maron BJ, Pelliccia A. The heart of trained athletes: cardiac remodeling and the risks of sports, including sudden death. Circulation. 2006;114(15):1633–44.
11 Kato TS, Noda A, Izawa H, Yamada A, Obata K, Nagata K, et al. Discrimination of nonobstructive hypertrophic cardiomyopathy from hypertensive left ventricular hypertrophy on the basis of strain rate imaging by tissue Doppler ultrasonography. Circulation. 2004;110(25):3808–14.
12 Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62(5):999–1006.
13 Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358(18):1899–908.
14 Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531–47.
15 Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, Fonarow GC, et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation. 2016;134(23):e579–e646.
16 Damy T, Costes B, Hagege AA, Donal E, Eicher JC, Slama M, et al. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J. 2016;37(23):1826–34.
17 Phelan D, Collier P, Thavendiranathan P, Popovic ZB, Hanna M, Plana JC, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart. 2012;98(19):1442–8.
18 Elliott PM. Hypertrophic Cardiomyopathy: Job Done or Work in Progress? J Am Coll Cardiol. 2016;67(12):1410–1.
19 Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129–200.
20 Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation. 1981;63(6):1285–8.
21 Sigwart U. Non-surgical myocardial reduction for hypertrophic obstructive cardiomyopathy. Lancet. 1995;346(8969):211–4.
22 Morrow AG, Reitz BA, Epstein SE, Henry WL, Conkle DM, Itscoitz SB, et al. Operative treatment in hypertrophic subaortic stenosis. Techniques, and the results of pre and postoperative assessments in 83 patients. Circulation. 1975;52(1):88–102.
23 Kim LK, Swaminathan RV, Looser P, Minutello RM, Wong SC, Bergman G, et al. Hospital Volume Outcomes After Septal Myectomy and Alcohol Septal Ablation for Treatment of Obstructive Hypertrophic Cardiomyopathy: US Nationwide Inpatient Database, 2003–2011. JAMA Cardiol. 2016;1(3):324–32.
24 Agarwal S, Tuzcu EM, Desai MY, Smedira N, Lever HM, Lytle BW, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010;55(8):823–34.
25 O’Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014;35(30):2010–20.
26 Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart. 2014;100(6):465–72.
27 Guttmann OP, Pavlou M, O’Mahony C, Monserrat L, Anastasakis A, Rapezzi C, et al. Prediction of thrombo-embolic risk in patients with hypertrophic cardiomyopathy (HCM Risk-CVA). Eur J Heart Fail. 2015;17(8):837–45.
28 Manolio TA, Baughman KL, Rodeheffer R, Pearson TA, Bristow JD, Michels VV, et al. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop. Am J Cardiol. 1992;69(17):1458–66.
29 Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Bohm M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850–8.
30 Biesbroek PS, Beek AM, Germans T, Niessen HW, van Rossum AC. Diagnosis of myocarditis: Current state and future perspectives. Int J Cardiol. 2015;191:211–9.
31 Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34(33):2636–48, 48a–48d.
32 LeWinter MM, Granzier HL. Titin is a major human disease gene. Circulation. 2013;127(8):938–44.
33 Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619–28.
34 Ottenheijm CA, van Hees HW, Heunks LM, Granzier H. Titin-based mechanosensing and signaling: role in diaphragm atrophy during unloading? Am J Physiol Lung Cell Mol Physiol. 2011;300(2):L161–6.
35 Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36(18):1123–35a.
36 Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. 2016;374(3):233–41.
37 Guzzo-Merello G, Cobo-Marcos M, Gallego-Delgado M, Garcia-Pavia P. Alcoholic cardiomyopathy. World J Cardiol. 2014;6(8):771–81.
38 Mangiafico S, Costello-Boerrigter LC, Andersen IA, Cataliotti A, Burnett JC, Jr. Neutral endopeptidase inhibition and the natriuretic peptide system: an evolving strategy in cardiovascular therapeutics. Eur Heart J. 2013;34(12):886–93c.
39 McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371(11):993–1004.
40 Kober L, Thune JJ, Nielsen JC, Haarbo J, Videbaek L, Korup E, et al. Defibrillator implantation in patients with nonischemic systolic heart failure. N Engl J Med. 2016;375(13):1221–30.
41 Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS: The Task Force for the management of atrial fibrillation of the European Society of Cardiology (ESC)Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESCEndorsed by the European Stroke Organisation (ESO). Eur Heart J. 2016.
42 Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60(8):705–15.
43 Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011;13(8):1077–109.
44 Ingles J, McGaughran J, Scuffham PA, Atherton J, Semsarian C. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart. 2012;98(8):625–30.
45 Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343(23):1688–96.
46 Waldmuller S, Erdmann J, Binner P, Gelbrich G, Pankuweit S, Geier C, et al. Novel correlations between the genotype and the phenotype of hypertrophic and dilated cardiomyopathy: results from the German Competence Network Heart Failure. Eur J Heart Fail. 2011;13(11):1185–92.
47 Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J. 2012;33(14):1724–33.
48 Helms AS, Day SM. Hypertrophic cardiomyopathy: single gene disease or complex trait? Eur Heart J. 2016;37(23):1823–5.