La prise en charge des pathologies cognitives du cerveau âgé est marquée par une évolution constante de ses conceptions physiopathologiques et des techniques de diagnostic. Ces nouvelles conceptions concernent la critériologie diagnostique et les perspectives thérapeutiques mais aussi une démarche pour intégrer ces connaissances en pratique quotidienne.
Introduction
L’évolution des connaissances sur les maladies conduisant à une démence, en particulier la maladie d’Alzheimer (MA), a permis de comprendre que les processus physiopathologiques sous-jacents, dont les causes sont multifactorielles, se développent progressivement sur une vingtaine d’années avant d’avoir une manifestation clinique. Celle-ci se traduit par des symptômes cognitifs et comportementaux évolutifs dont les phénotypes traduisent la topographie lésionnelle cérébrale. L’avènement des biomarqueurs accessibles en recherche, puis en pratique clinique a permis de faire évoluer les critères de ces maladies et d’appréhender le diagnostic étiologique in vivo en passant d’un raisonnement clinico-radiologique probabiliste vers un raisonnement clinico-biologique de quasi-certitude. L’ensemble de ces connaissances nous fait comprendre que le syndrome démentiel représente le stade ultime de ces maladies et que la principale raison de l’augmentation de sa prévalence est liée à l’allongement de l’espérance de vie. Dans la pratique courante, l’intérêt évident du diagnostic précoce des troubles cognitifs en lien avec une pathologie dégénérative repose sur la prévention, par une prise en charge symptomatique, des complications d’ordre médico-social qui émaillent l’évolution de ces maladies. Etre attentif à la plainte et aux troubles cognitifs du patient permet aussi de rechercher des diagnostics différentiels pour lesquels un retard diagnostique représentent une perte de chance pour le malade. L’utilisation des biomarqueurs chez les patients les plus jeunes ou lorsque les présentations cliniques sont atypiques ou associés à des signes psychiatriques permet d’éviter une errance diagnostique. Une action coordonnée des médecins de premiers recours et des centres mémoires, en étroite relation avec les professionnels médico-sociaux, est utile aux patients et à leurs familles. Enfin, la communauté médicale doit intégrer qu’elle a, comme pour les autres maladies chroniques d’organes, un rôle primordial pour la prévention primaire et le repérage des premiers symptômes, et qu’elle aura à moyen terme un rôle majeur pour le dépistage et l’orientation des sujets à risque vers les centres spécialisés pour des traitements innovants.
La maladie d’Alzheimer: évolution des connaissances
Epidémiologie
La MA est la principale cause de démence. Elle représente, compte tenu de l’augmentation de sa prévalence et de son incidence avec l’âge, un enjeu sanitaire et social majeur du fait de la dépendance qu’elle génère. Cette maladie cérébrale s’exprime, en général, après 65 ans même si des sujets plus jeunes peuvent être affectés. On a estimé, en 2015 dans le monde, que 47 millions de personnes présentaient une démence et que ce chiffre pourrait être multiplié par 3 en 2050 [1]. En Suisse, on estime que 144 000 personnes sont concernées [2]. La prévalence après 65 ans est de 5% et atteint 20% des plus de 85 ans [1]. Le sex ratio est de 2 femmes / 1 homme. Les comorbidités lésionnelles cérébrales sont fréquentes et augmentent après 80 ans. Elles sont très souvent vasculaires ou liées à d’autres processus dégénératifs [3]. Des facteurs de risques et des facteurs protecteurs ont été identifiés. On parle de facteurs de risque modifiables et non modifiables (âge, sexe, gènes). Les facteurs modifiables sont présentés dans le tableau 1. Certains ont un rôle spécifique dans la physiopathologie de la maladie. D’autres ont un rôle moins clair et peuvent avoir, notamment pour les facteurs protecteurs, un effet favorable sur les systèmes de compensation (réserve cognitive) de l’individu permettant à ce dernier de «lutter» contre le déclin. Plusieurs études ont observé une diminution de l’incidence de la démence ces dernières années. Les auteurs suggèrent, alors qu’il n’y a pas eu de traitements nouveaux, que les sujets de plus de 65 ans d’aujourd’hui diffèrent de ceux d’il y a 10 ans principalement par leur différence de mode de vie et d’accès aux soins, en particulier dans le domaine vasculaire [4].
Tableau 1: Facteurs de risque et facteurs protecteurs (inspiré de [1]).
Facteurs
Type
Niveau de preuve (+ à +++) et périodes de la vie significatives
Traumatisme cranio-cérébral
Risque
+++
Obésité
Risque
++ milieu de la vie (45–65)
Hypertension arterielle (HTA)
Risque
++ milieu de la vie (45–65)
Tabagisme actif
Risque
++
Diabète de type 2
Risque
++
Isolement social
Risque
+ tardif (>65)
Dépression
Risque
+ tardif (>65)
Troubles du sommeil
Risque
+ tardif (>65)
Dyslipidémie
Risque
+
Hypoacousie
Risque
+ milieu de la vie (45–65)
Niveau de scolarité secondaire et plus
Protecteur
+++
Activité physique régulière
Protecteur
++ milieu de vie (45–65)
Régime méditerranéen
Protecteur
++
Stimulation intellectuelle
Protecteur
++
Consommation modérée d’alcool
Protecteur
+
Engagement social
Protecteur
+
Physiopathologie
La MA est caractérisée par la présence anormale au sein du parenchyme cérébral de deux processus lésionnels. Il s’agit d’une part des plaques amyloïdes, liées à l’agrégation de dépôts extra-cellulaires de peptides amyloïdes dans leurs formes pathologiques (AB42 et AB40). D’autre part, il s’agit de lésions intra-cellulaires de dégénérescences neuro-fibrillaires (DNF) liées à l’accumulation de protéines TAU anormalement phosphorylées. Ces lésions sont connues depuis de nombreuses années mais la description de leur cinétique d’apparition est récente. L’avènement des techniques de neuro-imagerie et de neurobiologie a permis de construire, in vivo, un modèle dynamique de la MA [5]. En effet, il est maintenant admis que la MA est une maladie cérébrale chronique pour laquelle les lésions s’installent insidieusement durant une vingtaine d’années, avec d’abord l’amyloïdopathie et ensuite la TAUpathie. L’amyloïdopathie est un processus diffus, qui semble causer principalement une vulnérabilité cérébrale et qui favorise, directement ou indirectement l’apparition du processus neurodégénératif caractérisé par la pathologie TAU. Ce modèle semble être identique pour les formes sporadiques et les rares formes familiales de MA, c’est-à-dire celles dont l’âge de survenue est en général inférieur à 50 ans et dont le mode de transmission est de type autosomique dominant (AD).
La différence entre ces 2 formes est le mécanisme de l’amyloïdopathie. En effet, pour les formes familiales, l’accumulation du peptide amyloïde est liée à une hyperproduction alors que pour les formes sporadiques, cette accumulation serait liée à un défaut de clairance du peptide, le taux de production n’étant, a priori, pas différente des sujets sains [6]. Ce défaut serait principalement en lien avec une vasculopathie sous-jacente. Bien que cela fasse l’objet de débats, cette hypothèse est congruente avec les données épidémiologiques, en particulier à propos de facteurs de risques cérébro-vasculaires et du facteur de susceptibilité que représente le statut génétique APOE4. Des mécanismes inflammatoires sont aussi présents pour lesquels l’aspect primaire et/ou secondaire reste un sujet de réflexions ouvert.
Nouvelles conceptions diagnostiques et nouvelle terminologie d’abord pour la recherche
Jusqu’au début des années 2000, le diagnostic de MA reposait sur les critères élaborés au stade de démence. A la fin des années 1990, le concept de «mild cognitive impairment» (MCI ou trouble cognitif léger) a émergé. L’avantage de ce concept a été de montrer qu’il y avait un «état» transitionnel entre le vieillissement cognitif normal et la démence. Toutefois, les critères de MCI, basés sur des données cliniques, ont montré leurs limites par absence de spécificité, ce qui explique en partie la négativité des études médicamenteuses réalisées avec cette population. Dubois et al. [3, 7] ont fait évoluer les critères en identifiant, au sein des «MCI», les sujets ayant un stade qualifié de pré-démentiel de la MA. Il s’agit d’identifier les patients ayant un profil amnésique quasiment isolé grâce à un test psychométrique sensible et assez spécifique pour l’évaluation de la mémoire épisodique, type de mémoire altéré lors du dysfonctionnement de l’hippocampe, cible initiale de la MA dans sa forme typique. L’avènement des techniques biologiques et d’imagerie métabolique depuis le début des années 2000 a permis de les confronter aux données cliniques et radio-morphologiques et de se rendre compte que ces différents critères opérationnels, de type clinico-radiologique, utilisés jusqu’alors manquaient de spécificité. En effet, le symptôme clinique et l’atrophie cérébrale sont des marqueurs topographiques et des auteurs ont proposé d’intégrer les bio-marqueurs du processus pathologique dans le raisonnement étiologique. Ces biomarqueurs TAU et amyloïde sont accessibles in vivo dans le LCR et en imagerie métabolique à l’aide du PET scanner.
Ces nouvelles données suggèrent 2 stades de la MA:
– Le stade clinique associé à la positivité des 2 biomarqueurs. Ce stade clinique inclut les phases pré- et démentielle de la MA. En 2013, le DSM 5 a requalifié ces 2 phases en troubles neurocognitifs «mineurs» et «majeurs». Celles-ci sont définies de manière multi-dimensionnelle, mais peuvent se résumer par le fait de savoir s’il existe une perte d’autonomie dans la vie quotidienne (trouble majeur) ou non (trouble mineur).
– Le stade pré-clinique, identifié par la présence des 2 biomarqueurs TAU et amyloïde au niveau cérébral, définissant le début de la maladie au sens biologique, sans qu’aucun symptôme ne soit identifié. Il semble toutefois, qu’un certain nombre de sujets rapportent une plainte cognitive subjective en toute fin de ce stade et font l’objet de nombreuses recherches actuellement.
– Un 3ème stade encore plus précoce nommé «stade asymptomatique à risque de MA», stade où seule l’amyloïdopathie est objectivable.
L’intégration des biomarqueurs dans le raisonnement clinique a aussi permis d’identifier des phénotypes différents de la MA avec la forme typique où prédomine le syndrome amnésique et des formes atypiques inaugurées soit par un trouble progressif du langage, de la vision, ou du comportement de type frontal.
Cette évolution conceptuelle de la MA a été dictée par l’évolution des connaissances cliniques, physiopathologiques et des techniques biologiques et d’imagerie. Elle est aussi due aux leçons tirées des échecs successifs des essais médicamenteux réalisés depuis 2003.
En 2018, comment faire le diagnostic de MA en pratique quotidienne et pourquoi le faire?
Actuellement, il n’est pas recommandé de dépister la MA, c’est-à-dire de rechercher les lésions pathologiques dans les liquides ou les tissus biologiques alors que le sujet n’exprime aucune plainte ou troubles cognitifs. Bien que cette maladie soit grave et fréquente, plusieurs raisons expliquent cette position et la première est d’ordre éthique puisque nous n’avons aucun médicament curatif à notre disposition. D’autre part, nous n’avons pas de certitude à l’échelle individuelle sur la cinétique d’apparition des symptômes chez des sujets qui auraient un dépistage positif. En revanche, il est recommandé de porter un diagnostic chez un sujet qui exprime une plainte cognitive ou chez qui l’entourage ou le médecin traitant signalent un changement cognitif ou comportemental. Selon les recommandations européennes, ce diagnostic doit être porté au moment «opportun», c’est-à-dire au moment où le patient ou sa famille s’interrogent sur le fonctionnement cognitif ou le statut fonctionnel. Cela peut-être très précoce dans l’apparition des symptômes ou plus tardif.
Cette position est respectable mais nous pensons que la réalisation d’un diagnostic précoce est garante d’un meilleur plan de soin. Premièrement, tout trouble cognitif n’est pas synonyme de MA même à un âge avancé. Au sein des centres mémoire, le diagnostic de MA ne représente «que» 25% environ des cas examinés. Des diagnostics différentiels comme une dysthyroïdie, un processus expansif, une encéphalite auto-immune, une dépression, une autre maladie dégénérative, etc. sont autant d’étiologies possibles où un retard diagnostique est une perte de chance. Il faut toutefois noter que dans un certain nombre de cas, la plainte peut être isolée, sans troubles observés. Ce n’est pas synonyme de MA mais cela représente un facteur de risque, voire potentiellement un premier symptôme, surtout chez les sujets de plus de 55 ans, motivant un suivi chez ces patients.
Dans la pratique courante actuelle, l’intérêt du diagnostic précoce des troubles cognitifs en lien avec une pathologie dégénérative, vise à prévenir, par une prise en charge symptomatique, des complications d’ordre médico-social et juridique qui émaillent immanquablement l’évolution de ces maladies. Cela permet d’avoir le temps pour les explications à fournir au patient quant aux troubles qu’il présente et aussi pour le praticien d’appréhender les choix et les décisions du malade par rapport aux ressources existantes dans le plan de soins global que l’on peut envisager au gré de l’évolution possible. Ce diagnostic permet au patient de mieux anticiper les mesures à prendre dans le futur telles que les dispositions du droit de protection de l’adulte (directives anticipées, représentant thérapeutique, mandat pour causes d’inaptitude). Ces soins symptomatiques, médicamenteux et psycho-sociaux précoces, permettent aussi de différer l’apparition de certains symptômes en particulier comportementaux et la perte d’autonomie, «d’éduquer» l’entourage familial à la communication et la gestion des symptômes et d’accompagner le patient et sa famille dans une situation qui génère une rupture de la dynamique familiale, souvent traumatisante. Ces aides précoces permettent aussi de réduire les coûts économiques, pour la famille et la société, en retardant l’institutionnalisation.
Entendre la plainte
L’analyse de la plainte a son importance, en particulier s’il s’agit d’oublis d’évènements récents alors que les faits anciens sont bien rappelés, de répétitions de questions, d’oublis de rendez-vous importants. La notion de changement cognitif par rapport à l’état antérieur mais aussi la notion d’effort intellectuel pour réaliser des tâches qui étaient aisées quelques mois auparavant doivent retenir l’attention. Il faut aussi retenir qu’une plainte cognitive récente, c’est-à-dire inférieure à 3 ans, est suspecte et qu’elle mérite une évaluation exhaustive même si les tests de repérage sont dans les normes. Une symptomatologie dépressive est fréquente mais elle est souvent confondue avec l’apathie, qui est présente dans ¾ des cas, dès les stades précoces. L’anxiété est aussi présente, souvent liée aux capacités du patient à se rendre compte de ses troubles et représente un état pré-morbide fréquent. Certains sujets présentent d’emblée une anosognosie mais cela est souvent plus tardif, et on ne doit pas la confondre avec un mécanisme de déni des troubles qui représentent une défense psychique face à la pathologie.
Identifier un trouble cognitif
Ce type de plainte associée ou non à quelques «drapeaux rouges» comme les facteurs de risque vasculaires, la symptomatologie dépressive, les antécédents familiaux de MA doit être investiguée par le médecin de premier recours avec des outils de «repérage», associé à l’examen somatique. Ces outils sont des tests psychométriques validés qui permettent de se faire une impression sur le fonctionnement cognitif de l’individu. Ils permettent d’évaluer rapidement l’ensemble des fonctions cognitives. On citera par exemple le «Mini-Mental State Examination» (MMSE) ou le «Montreal Cognitive Assessment» (MoCA). Ces tests ne permettent pas de porter un diagnostic mais ils permettent d’identifier la sévérité des troubles cognitifs. Il faut tenir compte aussi de nombreux paramètres et savoir qu’ils ont un effet plafond chez les individus de haut niveau d’éducation.
Argumenter le diagnostic
Imagerie cérébrale
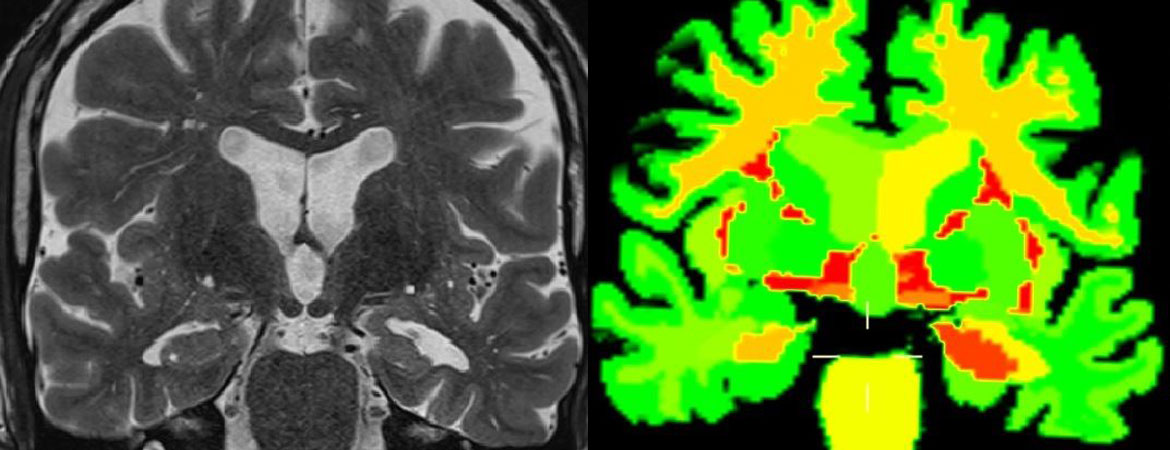
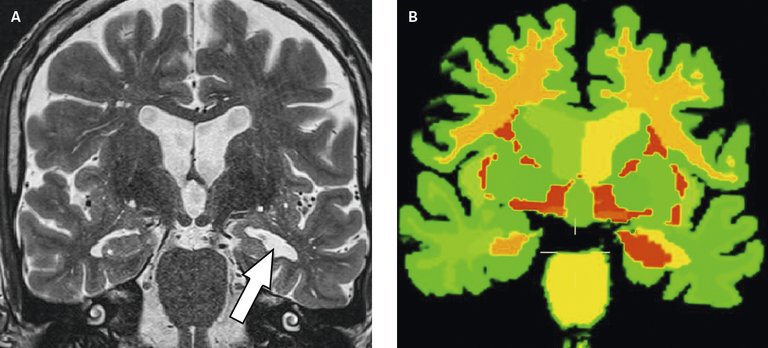
Les examens d’imagerie permettent d’orienter le diagnostic. On distingue l’imagerie morphologique (IRM et Scanner) et l’imagerie fonctionnelle (PET et SPECT). Il est recommandé de réaliser une imagerie morphologique pour tout bilan diagnostique de déclin cognitif. L’imagerie est aussi obligatoire en cas d’aggravation très rapide ou atypique chez tout patient et même s’il présente un syndrome démentiel préalablement connu. L’IRM est l’examen de choix car elle permet entre autres une étude précise de la morphologie (volume) des régions cérébrales (fig. 1). Par conséquent, l’IRM permet d’argumenter en faveur d’un diagnostic positif probabiliste d’une maladie dégénérative ou d’identifier d’autres causes telles les lésions tumorales ou des lésions vasculaires. Dans la forme typique de la MA, une atrophie hippocampique sera recherchée. Il existe une corrélation positive entre les symptômes et l’atrophie des réseaux anatomo-fonctionnels impliqués. La modification structurale est tardive dans l’évolution du processus dégénératif et peut être absente au début des symptômes.
Figure 1: A) IRM Coupe coronale séquence FLAIR montrant une atrophie du cortex hippocampique gauche (Courtesy of Dept Radiologie CHUV & Siemens/CHUV/EPFL). B) Même données IRM après traitement par logiciel MorphoBox® (Siemens) permettant automatiquement la segmentation en régions anatomiques et la quantification de leur volume, ainsi que la comparaison de cette mesure à une valeur de référence régionale; le codage couleur montre pour les couleurs orange à rouge une importante déviation par rapport à la norme (rouge: valeur de Z score ≥3). (Courtesy of Dept. Radiologie CHUV & Siemens/CHUV/EPFL); l’atrophie hippocampique gauche est particulièrement bien visualisée par cette quantification mais celle-ci révèle également une diminution de volume très significative dans la substance blanche des deux hémisphères.
Biologie
Un bilan biologique est réalisé pour éliminer des causes classiques de pathologies non-neurologiques se manifestant par des troubles cognitifs ou pour rechercher des comorbidités fréquentes comme les carences vitaminiques B12 et B9. Dans les situations les plus fréquentes, il est recommandé de réaliser hémogramme, VS, ionogramme avec calcémie et TSH. En cas d’anomalies, il est recommandé de le corriger et de réévaluer les fonctions cognitives ensuite. En fonction du contexte, les sérologies syphilis ou VIH sont réalisées.
Confirmation diagnostique par le spécialiste et rôle des centres mémoire
Lorsque le médecin de premier recours évoque l’hypothèse d’un trouble cognitif acquis, suspect d’être en lien avec une pathologie organique cérébrale, l’avis d’un spécialiste est requis. Pour les sujets de plus de 75 ans avec perte d’autonomie, l’orientation vers un gériatre formé aux pathologies démentielles est préconisée car avec l’âge avancé, il est fréquent qu’il y ait de nombreuses comorbidités d’organes, sensorielles ou fonctionnelles. L’expertise clinique d’un psychiatre a son intérêt lorsqu’il y a une symptomatologie thymique ou comportementale inaugurale pour juger d’une cause psychiatrique éventuelle et orienter au mieux le traitement psychotrope. Les pathologies dégénératives acquises du cerveau peuvent avoir différentes présentations cognitives ou comportementales et varier aussi en fonction du terrain sur lequel elles surviennent. Les neurologues orienteront au mieux la stratégie d’examens complémentaires pour spécifier le diagnostic étiologique. Au-delà de ces spécialisations médicales, l’expertise pluridisciplinaire a toute son importance et c’est la raison pour laquelle des centres mémoire ont été mis en place dans de nombreux sites hospitaliers ou non. Ces centres sont composés d’équipes médicales pluridisciplinaires (gériatres, neurologues et psychiatres) et interprofessionnelles (neuropsychologues, psychologues, infirmières, assistantes sociales, secrétaires). Le centre mémoire peut, du fait de ses compétences, être un recours pour le médecin traitant et aussi être un recours de 2ème ligne pour les spécialistes.
Lorsque la symptomatologie est peu marquée, dans les stades légers des troubles (MMSE >24/30 ou MOCA >20/30), il est nécessaire d’affiner l’évaluation cognitive faite par le médecin à l’aide d’un bilan exhaustif réalisé par des neuropsychologues. Ce bilan permet d’apprécier les performances dans les différents domaines cognitifs à l’aide de tests validés et normés. Ce bilan permet d’identifier un «profil cognitif» précis et de faire un diagnostic syndromique. La réalisation de ce diagnostic permet en premier lieu d’aider le clinicien à faire un diagnostic étiologique à partir des critères opérationnels, en tenant compte des éléments anamnestiques, cliniques et para-cliniques, mais aussi d’identifier les fonctions cognitives efficientes dans l’objectif d’aider le patient à compenser au mieux ses difficultés dans la vie quotidienne. Avec ces éléments, le diagnostic étiologique repose sur un faisceau d’arguments clinico-radiologiques et est par conséquent probabiliste du vivant du patient, alors que le diagnostic de certitude reste anatomopathologique. Chez des sujets jeunes ou dans les cas complexes, un SPECT ou un PET FDG peuvent apporter des arguments en mettant en évidence une hypoperfusion ou un hypométabolisme, particulièrement si aucune atrophie n’est observée à l’IRM. Ce type d’examen apporte un argument lui aussi topographique. Le raisonnement diagnostique probabiliste est suffisant chez les sujets de plus de 65 ans ayant une présentation clinique typique dans l’objectif d’une prise en charge symptomatique. En effet, le «gain» diagnostique influe peu sur le plan de soin dans ces situations. En revanche, chez les sujets plus jeunes, dans l’hypothèse d’une MA, il est recommandé d’avoir une preuve biologique, accessible via l’études des biomarqueurs TAU et amyloïdes dans le LCR. Cela permet de porter un diagnostic avec un niveau de preuve élevé. L’intérêt est lié à la recherche de la cause de la maladie qui peut être génétique, de transmission autosomique dominante, en particulier chez les sujets de moins de 50 ans ou lorsque 2 cas documentés sont survenus dans une même famille avant 65 ans. D’autres méthodes non invasives ont été développées avec le PET et les ligands amyloïdes. Cela apporte la preuve d’une amyloïdopathie et son intérêt réside dans sa très bonne valeur prédictive négative. Il est utilisé surtout en recherche.
Avoir connaissance de la négativité des biomarqueurs permet d’explorer d’autres hypothèses dégénératives ou non et d’effectuer d’autres types d’examens en fonction du contexte (EEG, bilan auto-immun, recherche de maladies métaboliques, etc.). Les causes métaboliques sont les plus fréquentes avant l’âge de 35 ans mais après, les causes dégénératives deviennent les plus fréquentes.
Plan de soins ou «care today, cure tomorrow»
Il n’existe pas de traitement curatif de la MA. En revanche, comme pour beaucoup d’autres maladies chroniques d’organes, il existe des médicaments à visée symptomatique et des stratégies de prise en charge fonctionnelle. La MA est une maladie qui ne se guérit pas mais est une maladie qui se soigne.
Le plan de soins débute dès l’annonce diagnostique. Cette annonce représente un temps fondamental de la prise en charge et doit être réalisée dans le cadre d’une consultation de restitution dédiée. Les informations sur le contenu de ce qui a été formulé doivent être échangées entre le médecin spécialiste et le médecin de premier recours, ce afin de maintenir l’alliance thérapeutique. Dans les situations d’emblée plus sévères ou lorsque l’anosognosie est marquée, cette annonce est faite principalement à la personne la plus proche ayant le rôle d’aidant naturel dans l’entourage.
Concernant le plan de soins, il comprend 2 axes [11]:
– Un traitement médicamenteux symptomatique par inhibiteurs de choline-estérases est indiqué dans les formes légères à modérées; la mémantine est utilisée pour des formes plus sévères. Ces médicaments ont un effet modeste sur la cognition et sur l’impression clinique globale. L’intérêt de leur prescription existe s’il n’y a pas de contre-indications, de risque d’interactions ou de survenue d’effets indésirables. Les indications sont la démence d’Alzheimer ou la démence d’Alzheimer avec composante vasculaire. Des données récentes issues de l’étude DOMINO montrent un ralentissement du déclin chez les patients traités, au stade modéré à sévère de la maladie et une précipitation de l’entrée en institution en cas d’arrêt de traitement [12, 13]. Concernant la démence associée à la maladie de Parkinson et la démence à corps de Lewy, seule la rivastigmine a montré des preuves d’efficacité.
– La prise en charge psycho-sociale repose sur les aides à mettre en place. Elles ont pour objectif de maintenir le niveau d’autonomie du patient, de le protéger du fait de sa vulnérabilité et de ses potentielles difficultés de discernement et d’éviter un épuisement de l’aidant. Un suivi dans sa globalité est proposé et une attention particulière est aussi portée aux différentes comorbidités, souvent moins bien diagnostiquées ou prises en charge chez ces patients.
Prévention primaire et secondaire
Depuis les résultats des études médicamenteuses de la fin des années 1990 et début 2000, aucune nouveauté thérapeutique n’a été mise à disposition. Pourtant, plusieurs études ont montré une diminution significative de l’incidence des démences dans des cohortes actuelles de sujets de plus de 65 ans par rapport à celles de sujets de même tranche d’âge suivis 10 ans auparavant. La prévalence reste toutefois importante du fait de l’augmentation de la population et de l’espérance de vie. Ces données suggèrent que la démence est accessible à la prévention. Tenant compte des données des études épidémiologiques, les facteurs en cause dans ces différences entre les 2 périodes semblent être un plus haut niveau d’éducation et une meilleure prise en charge des facteurs de risque vasculaires. A la lumière de ces constatations, on identifie des facteurs de risque modifiables et non modifiables (âge, genre, génotype). Les facteurs de risque modifiables connus à ce jour sont ceux qui ont été présentés dans le tableau 1. Ils ont un impact significatif à différentes périodes de la vie et leur identification et prise en charge optimale pourraient avoir un effet significatif sur le délai d’apparition de la maladie. Certains de ces facteurs de risque sont impliqués dans la physiopathologie neurodégénérative, d’autres agissent sur les systèmes de compensation cognitive. Quelques études ont montré des effets positifs de protocoles d’intervention non médicamenteuse visant à corriger certains facteurs de risque [14].
Le cas des démences non-Alzheimer
D’autres maladies neurodégénératives que la MA ont une présentation inaugurale et principale de type cognitif et/ou comportemental. Les causes vasculaires secondaires à une macro ou micro-angiopathie représentent probablement la cause la plus fréquente de démence après la MA et les deux processus s’intriquent d’ailleurs fréquemment surtout chez les sujets âgés. La maladie à corps de Lewy associant des signes cognitifs aux symptômes parkinsoniens et psychiatriques est fréquente et sous-diagnostiquée. Les hallucinations visuelles sont précoces. Les différents syndromes de démences fronto-temporale (DFT) se manifestent principalement par des troubles comportementaux et sont souvent confondus avec des pathologies psychiatriques. D’autres pathologies telles que les syndromes corticaux basaux ou la paralysie supra-nucléaire progressive sont plus rares. La prise en charge psycho-sociale globale est souvent similaire que celle de la MA, mais suivant le contexte, elle peut varier dans ses modalités spécifiques de prise en charge; ces spécificités justifient d’autant plus le recours à un centre expert pour optimiser les plans de soins et informer au mieux les patients et leurs familles.
Conclusions et perspectives de recherches thérapeutiques
Les promesses attendues des recherches concernant des traitements «disease-modifyer» ne se sont pas encore concrétisées. Certes, l’absence d’effets chez l’homme des thérapeutiques efficaces sur les modèles animaux de la maladie est en partie lié aux imperfections de ces modèles. Diverses études (bapineuzumab et solanézumab) ont été négatives pour leurs critères principaux de jugement à savoir l’amélioration de la cognition et le maintien de l’autonomie. Cependant, cette absence d’effet est sans doute liée aux patients qui ont été recrutés pour ces études. Ces études ont eu le défaut d’inclure des patients qui soit présentaient un stade trop avancé dans le processus dégénératif soit ne correspondaient pas au diagnostic de MA. Pour autant, l’observation contre placebo d’effet biologique de ces molécules innovantes ainsi que leur relativement bonne tolérance incite à poursuivre dans cette voie tout en explorant d’autres pistes [15]; ainsi l’état d’esprit de la communauté médico-scientifique a évolué vers l’idée de prévenir la MA d’une part et de prévenir la démence due à la MA d’autre part. Ces pistes intègrent donc la notion de prévention mais aussi de dépistage et de diagnostic précoce.
La communauté médicale doit être informée de cette évolution et doit pouvoir répondre aux patients qui désirent accéder à l’innovation thérapeutique. C’est la raison pour laquelle, il nous semble essentiel que les patients qui expriment une plainte cognitive ou un trouble cognitif mineur puissent avoir accès à l’innovation thérapeutique dans le respect des bonnes pratiques cliniques et de l’éthique de la recherche clinique. Nous considérons que le défi de la MA est semblable à celui du VIH dans les années 1980 et que ce dernier a pu être relevé, même s’il y a encore des progrès à accomplir, grâce à la synergie des soins de premiers recours et des centres experts, mais aussi grâce à l’implication des associations de patients et de familles de patients. Comme pour le HIV ou les cancers, les avancées thérapeutiques ont été conditionnées par la précocité et la fiabilité des diagnostics et la recherche [16]. L’enjeu est tel pour notre société que l’ensemble des acteurs de santé et de sciences doit s’unir pour ce projet et que chacun y apporte sa contribution.
L’essentiel pour la pratique
• La Maladie d’Alzheimer (MA) est une maladie cérébrale chronique de cause multi-factorielle qui débute au milieu de la vie, qui s’exprime en général après 65 ans et qui n’est pas une conséquence inéluctable du vieillissement.
• Le DSM V remplace par «trouble neurocognitif majeur» le terme de «démence» et «mild cognitive impairment» (MCI) par «trouble neurocognitif mineur».
• La plainte cognitive est un motif fréquent de consultation en soin de premier recours. Ce n’est pas parce que c’est fréquent que c’est banal. La MA est une maladie fréquente, surtout après 65 ans.
• Evolution clinico-biologique des critères diagnostiques permettant un diagnostic précoce et fiable du vivant du patient.
• Perspectives thérapeutiques ciblées; avec la perspective d’une prise en charge de type P4: Préventive, participative, personnalisée, prédictive.
Disclosure statement
Les auteurs n’ont pas déclaré des obligations financières ou personnelles en rapport avec l’article soumis.
Correspondance
Prof. Dr méd. Jean-François Démonet Centre Leenaards de la Mémoire Centre hospitalier universitaire vaudois (CHUV) Rue du Bugnon 46 CH-1011 Lausanne Jean-Francois.Demonet[at]chuv.ch
Références
1 Livingston G, et al. Dementia prevention, intervention, and care. Lancet. 2017 Jul 19. pii: S0140 6736(17)31363-6.
3 Dubois B, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014 Jun;13(6):614–29.
4 Matthews FE, et al. A two decade dementia incidence comparison from the Cognitive Function and Ageing Studies I and II. Nat Commun. 2016 Apr 19;7:11398.
5 Jack CR Jr, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013 Feb;12(2):207–16.
6 Mawuenyega KG, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010 Dec 24;330(6012):1774.
7 Dubois B, et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016 Mar;12(3):292–323.
10 Annoni JM, Chouiter L, Démonet JF. Rev Med Suisse. Troubles cognitifs liés au vieillissement: évolution récente des concepts et stratégies diagnostiques . 2016 Apr 20;12(515):774–9.
11 Daher O, Nguyen S, Smith C, Büla C, Démonet JF. Les pathologies démentielles: Prise en charge et prévention. Rev Med Suisse. 2016 Apr 20;12(515):799–802.
12 Howard R, et al. Nursing home placement in the Donepezil and Memantine in Moderate to Severe Alzheimer’s Disease (DOMINO-AD) trial: secondary and post-hoc analyses. Lancet Neurol. 2015 Dec;14(12):1171–81.
13 Knapp M et al. Cost-effectiveness of donepezil and memantine in moderate to severe Alzheimer’s disease (the DOMINO-AD trial). Int J Geriatr Psychiatry. 2017 Dec;32(12):1205–16.
14 Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015 Jun 6;385(9984):2255–63.
15 Abbott A, Dolgin E. Failed Alzheimer’s trial does not kill leading theory of disease. Nature. 2016 Nov 23;540(7631):15–6.
16 Frisoni GB, Boccardi M, Barkhof F, Blennow K, Cappa S, Chiotis K, Démonet JF, et al. Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol. 2017 Aug;16(8):661–76.