

Publié le 28.08.2019
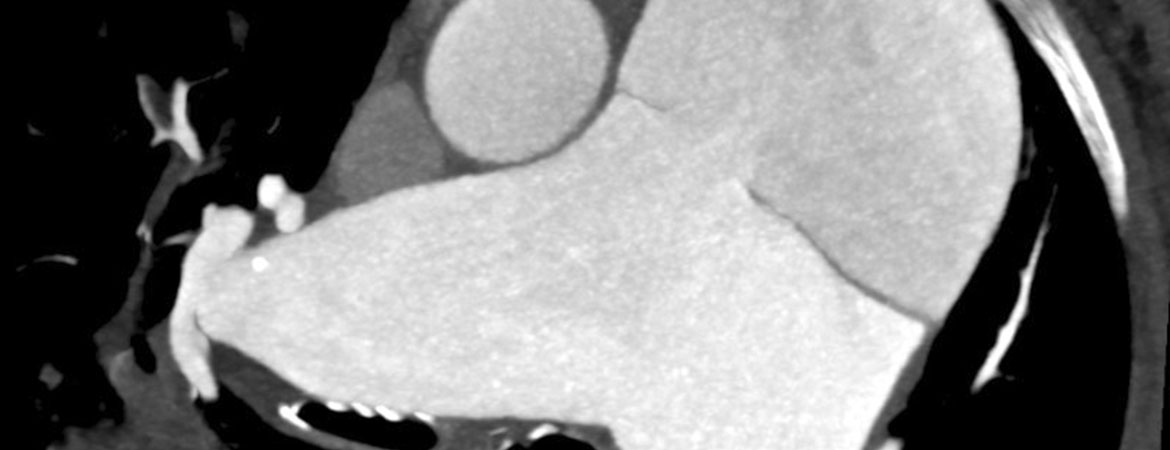
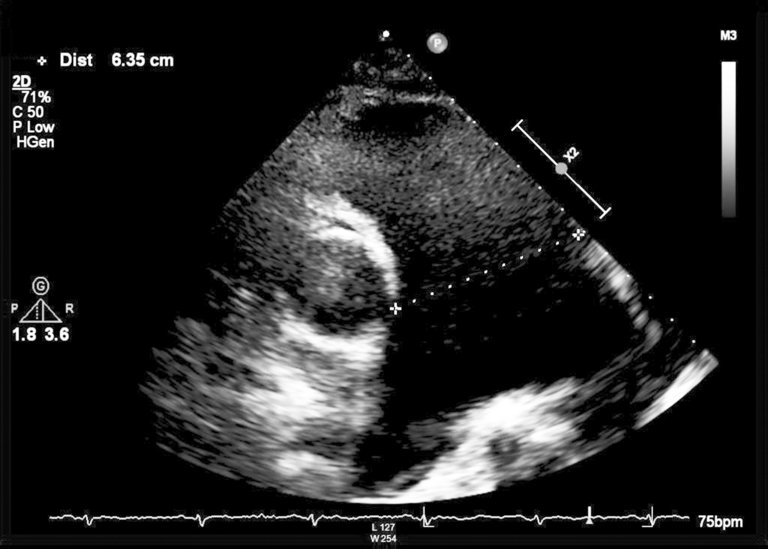
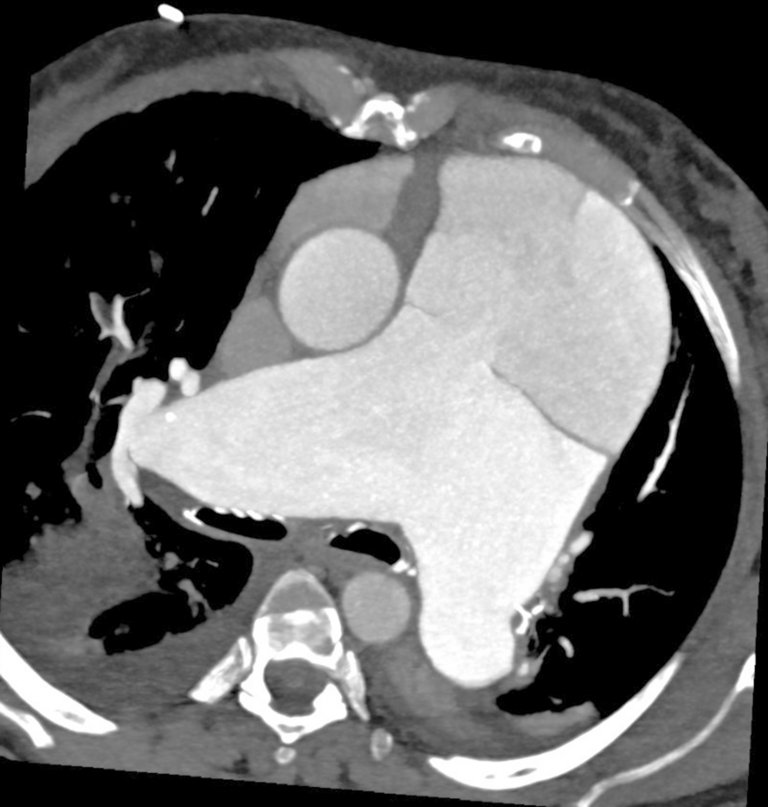
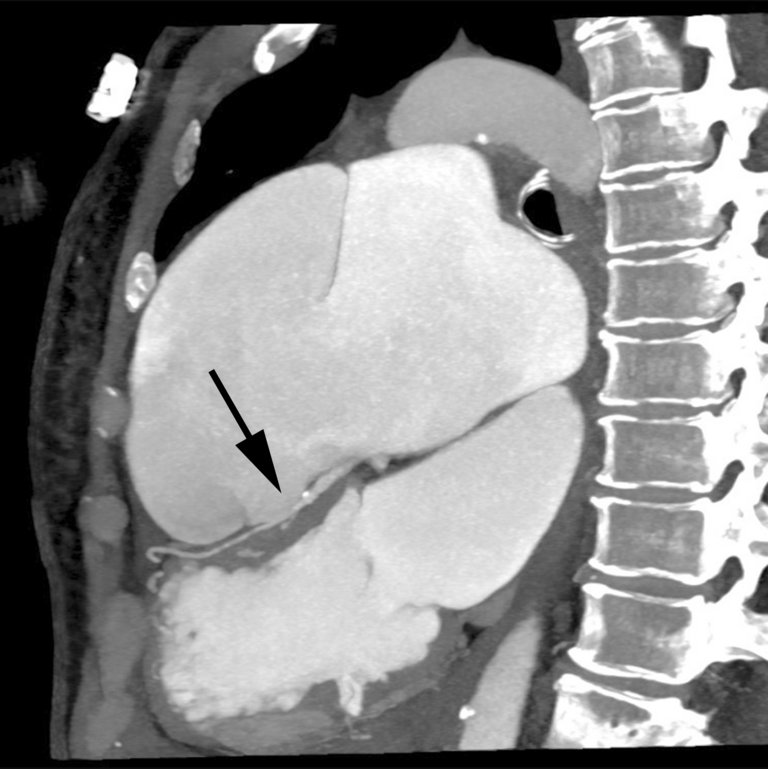
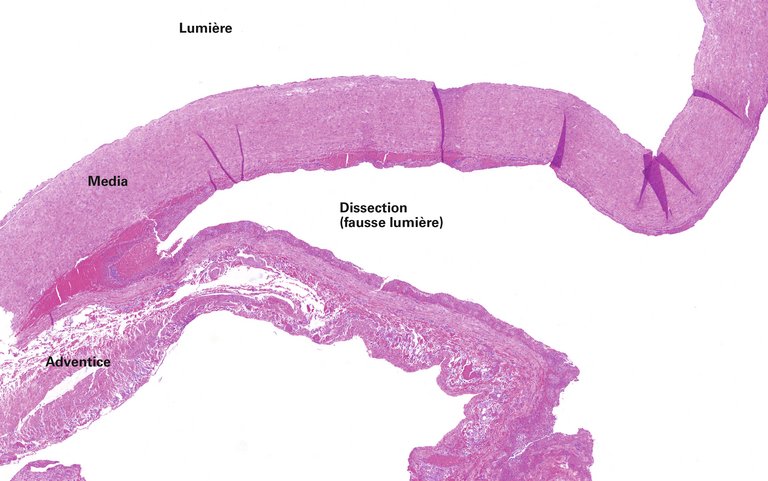
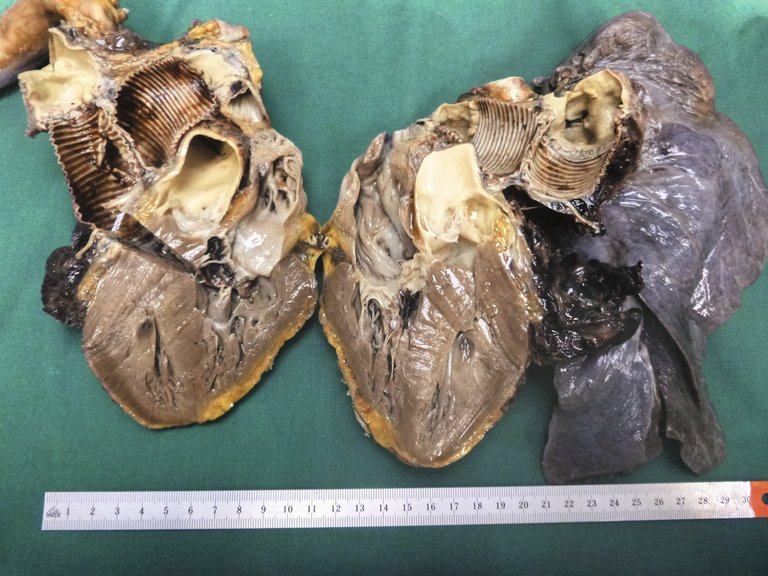
Une patiente de 65 ans atteinte d’une hypertension artérielle pulmonaire idiopathique vasoréactive connue depuis de nombreuses années, s’est présentée aux urgences avec une dyspnée aiguë et des douleurs thoraciques.

Publié sous la licence du droit d'auteur.
"Attribution - Non-Commercial - NoDerivatives 4.0"
Pas de réutilisation commerciale sans autorisation..
See: emh.ch/en/emh/rights-and-licences/
