a Medizinische Onkologie und Hämatologie, Kantonsspital, Winterthur; b Hämatologie und Center of Excellence of the European Competence Network on Mastocytosis, Luzerner Kantonsspital, Luzern; c Pathologie und Molekularpathologie, UniversitätsSpital, Zürich; d Pathologie, Kantonsspital, Winterthur; e Allergologie/Dermatologie, Kantonsspital, Winterthur
Dans cet article de revue, nous décrivons l’importance du diagnostic et du traitement d’une mastocytose systémique en nous appuyant sur un cas éloquent.
Evaluation des syncopes et anomalie cutanée – un cas pour une consultation d’hématologie?
Nous rapportons le cas d’un patient âgé de 30 ans qui a été retrouvé aréactif dans sa voiture sur le bord de route un jour d’été. A l’arrivée du service de secours, il y avait une suspicion clinique de crise convulsive, et le compte-rendu du service de secours indiquait en outre une anomalie cutanée avec des efflorescences ne disparaissant pas à la pression. Dans la salle de réanimation, le patient était hypotendu, normocarde, afébrile, normalement oxygéné et normoglycémique. Sur le plan clinique, il présentait un score de Glasgow (Glasgow Coma Scale) de 9 points (ouverture des yeux sur demande, pas réponse verbale, défense ciblée en réaction à la stimulation douloureuse). Par la suite, le patient est vite redevenu lucide et il était orienté sur le plan personnel, mais toujours désorienté temporellement et spatialement. Il a rapporté une sensation de malaise lors de la conduite, mais ne se souvenait pas des autres événements. L’imagerie par résonance magnétique (IRM) crânienne et l’analyse du liquide céphalo-rachidien n’ont pas présenté d’anomalie. Pendant l’hospitalisation qui a suivi et un séjour à la clinique d’épilepsie, le diagnostic de suspicion d’épilepsie du lobe temporal a été posé en raison d’un foyer temporal irritatif à l’électroencéphalogramme (EEG). Depuis lors, le patient recevait un traitement antiépileptique.
A l’initiative de la mère du patient qui avait fait des recherches au sujet des symptômes sur internet, le patient a été adressé à la consultation d’hématologie quelques mois après l’événement décrit plus haut. Le patient a rapporté avoir été victime à plusieurs reprises depuis 5 ans d’épisodes caractérisés par une sensation de chaleur, des rougeurs cutanées, une sensation de malaise, des fourmillements dans tout le corps, une dyspnée, des palpitations, des nausées et des douleurs abdominales. Un facteur déclenchant clair n’a pas pu être déterminé; une fois, les symptômes étaient apparus lors de l’application de crème solaire. Une perte de conscience n’était jamais survenue auparavant. Depuis sa jeunesse, le patient présentait en outre des altérations cutanées persistantes de couleur rouge-brune, avant tout sur le haut du corps. Les examens cardiologiques et dermatologiques réalisés il y a quelques années n’avaient pas montré de résultats expliquant les symptômes.
Le patient était au demeurant en bonne santé, sans aucun symptôme entre les épisodes mentionnés, il ne prenait pas de médicaments et il travaillait dans le domaine des transports publics.
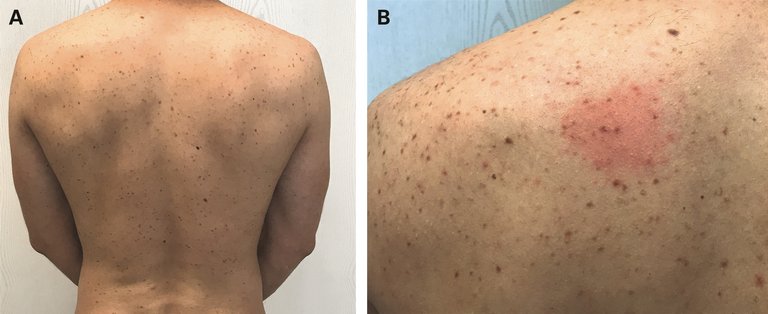
Sur le plan clinique, la seule anomalie constatée actuellement était des macules ovalaires de couleur rouge-brune disséminées, avant tout sur le dos. Après un bref frottement de la peau, une rougeur importante et un léger gonflement survenaient (fig. 1).
Figure 1: Macules ovalaires de couleur rouge-brune sur le dos (A). Gonflement urticarien des lésions après un court frottement (signe de Darier, B).
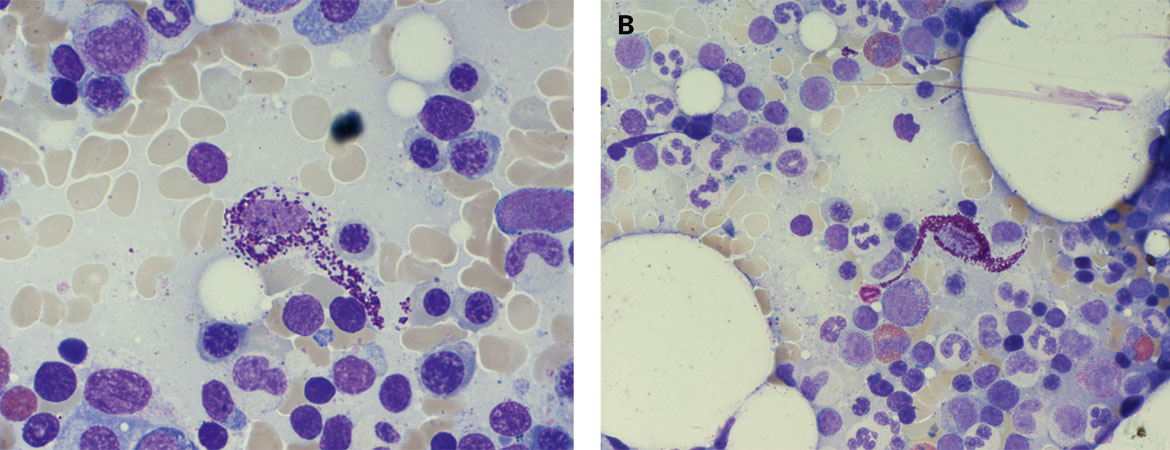
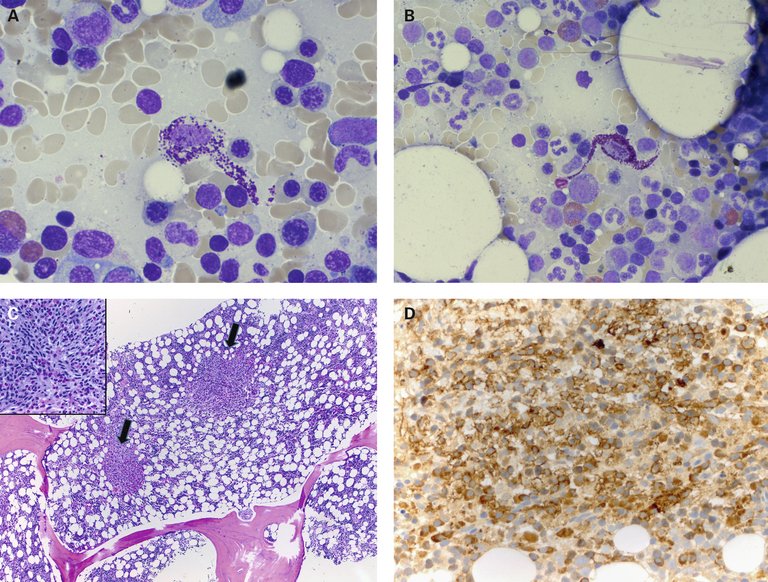
Les analyses de laboratoire ont révélé un hémogramme différentiel microscopique normal. En raison du diagnostic de suspicion clinique déjà posé, la tryptase sérique basale a été déterminée. Elle était considérablement accrue, de 52 µg/l (norme <11 µg/l). Ainsi, l’examen clinique et les analyses de laboratoire ont corroboré la suspicion d’une mastocytose systémique avec atteinte cutanée, qui a été confirmée par la biopsie cutanée. Afin d’approfondir l’évaluation et de compléter le diagnostic, une aspiration et biopsie medullaires ont été effectuées, révélant des agrégats disséminés de mastocytes atypiques. Sur le plan morphologique, les mastocytes présentaient une granulation cytoplasmique nettement atténuée et un aspect fusiforme atypique. Au demeurant, il y avait une myélopoïèse affichant une maturation normale, avec un nombre de cellules légèrement accru ainsi qu’une éosinophilie (fig. 2).
Figure 2: A) Mastocyte atypique fortement hypogranulaire (aspiration de moelle osseuse, coloration de May-Grünwald Giemsa, x500). B) Mastocyte atypique fusiforme (aspiration de moelle osseuse, coloration de May-Grünwald Giemsa, x500). C) Prolifération néoplasique nodulaire de mastocytes fusiformes (flèche) avec granulocytes éosinophiles mélangés (biopsie de moelle osseuse, coloration HE ×50, inset ×400). D) Expression immunohistochimique du CD117 des mastocytes néoplasiques (biopsie de moelle osseuse, ×400).
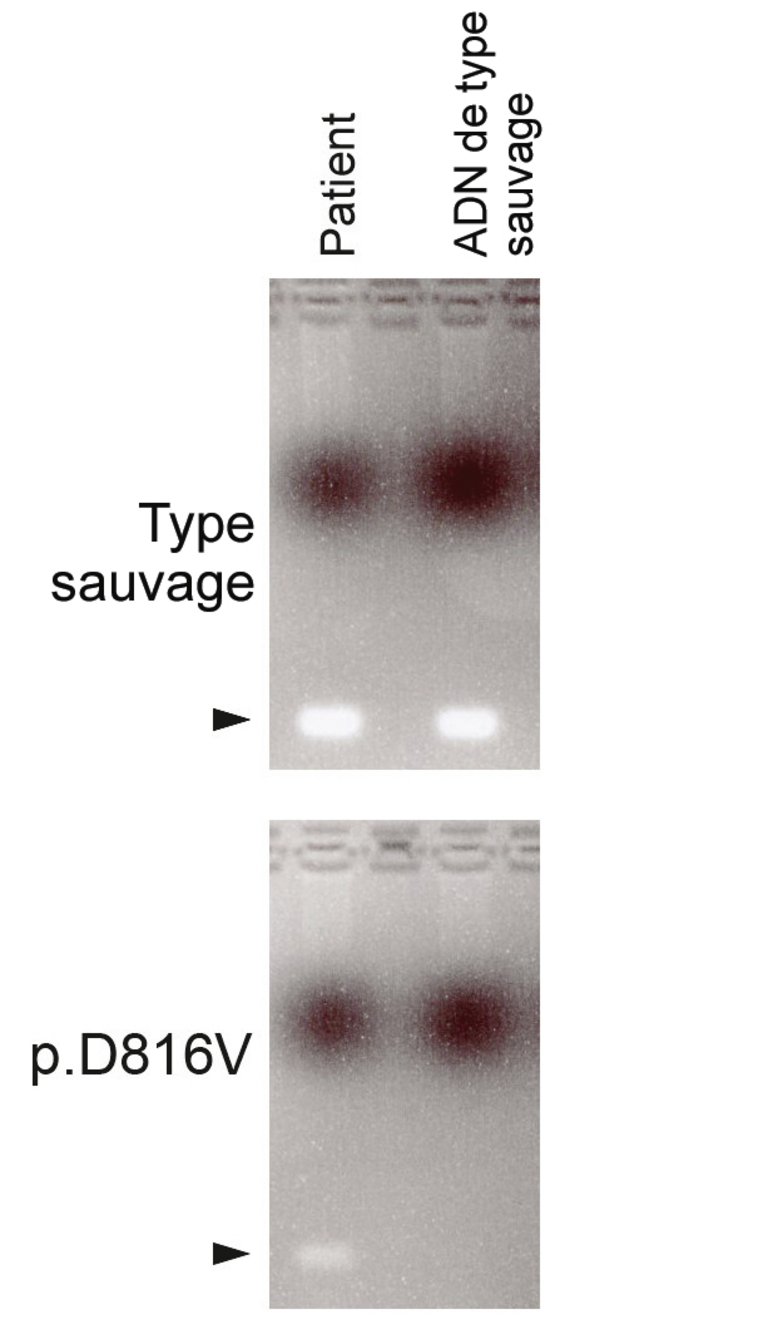
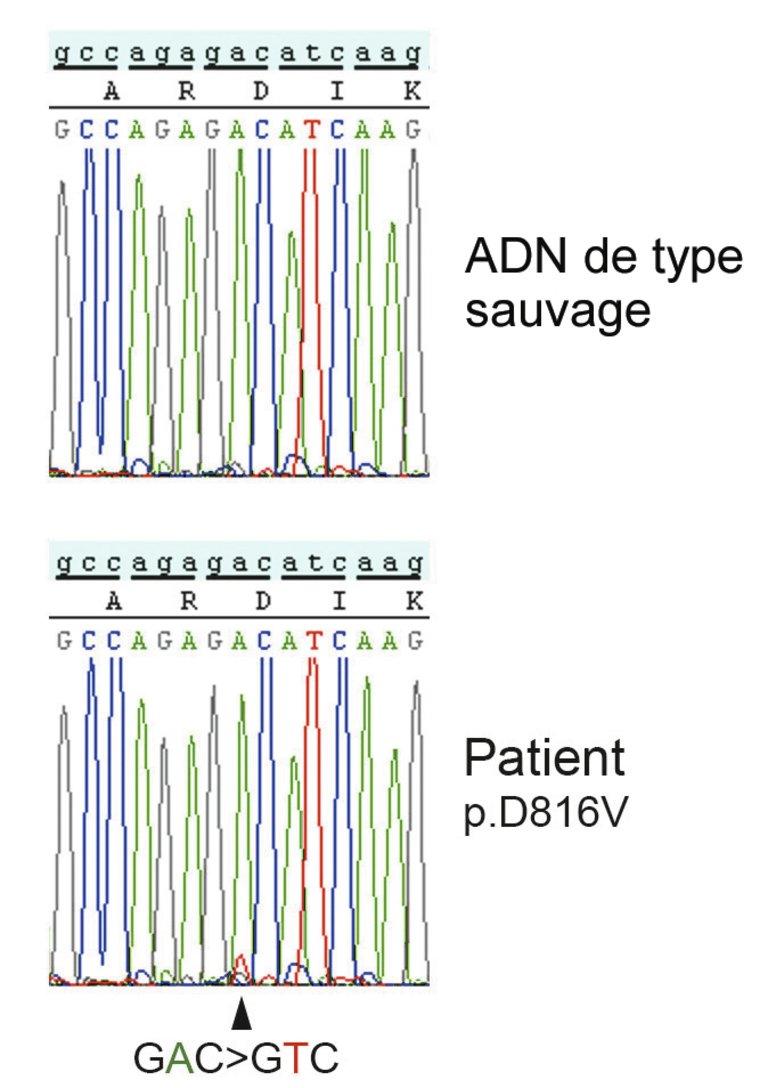
L’analyse FACS («fluorescence-activated cell sorting», cytométrie en flux) a permis de mettre en évidence une expression aberrante de l’antigène CD25 (sous-unité alpha du récepteur de l’interleukine-2), c’est-à-dire une expression non présente sur les mastocytes normaux. L’analyse de pathologie moléculaire a montré une mutation ponctuelle sur le codon 816 du gène KIT, avec une substitution de l’acide aminé aspartate par la valine (KITp.D816V) (fig. 3–4).
Figure 3: PCR allèle-spécifique. En haut: Amorce de KIT spécifique au type sauvage: dans les deux colonnes (patient et contrôle de type sauvage), une amplification du KIT de type sauvage est observée. En bas: Amorce de KIT spécifique à la mutation p.D816V: dans la colonne du patient, contrairement à la colonne de l’ADN contrôle de type sauvage, une bande est mise en évidence.
Figure 4: Séquençage Sanger. En haut: ADN contrôle de type sauvage. En bas: ADN du patient avec mise en évidence d’une faible proportion d’ADN avec la mutation ponctuelle GAC en GTC sur le codon 816, qui entraîne la substitution de l’acide aminé aspartate (D) par la valine (V) dans la protéine KIT .
L’échographie abdominale a montré une splénomégalie massive avec un diamètre de la rate de 13,4 cm et un volume de 425 ml. A l’ostéodensitométrie, une ostéopénie du rachis lombaire a été objectivée, avec une densité osseuse au demeurant normale.
En résumé, le diagnostic de mastocytose systémique indolente en tant que cause des symptômes récidivants a été posé.
Introduction
Les mastocytes sont produits dans la moelle osseuse à partir de cellules souches hématopoïétiques pluripotentes et ils maturent dans le tissu conjonctif périphérique. Le développement des mastocytes est régulé par le biais du récepteur de la tyrosine kinase KIT (une protéine membranaire; synonyme: CD117 ou récepteur du facteur de croissance des cellules souches) et est activé par son ligand, le facteur de croissance des cellules souches [1]. La dégranulation mastocytaire peut d’une part être médiée par les IgE (spécifiques aux allergènes, allergie de type immédiat), et d’autre part avoir lieu indépendamment des IgE (par exemple complément/ récepteurs du complément; quinolones au niveau du MRGPR [Mas-related G protein-coupled receptor]; lidocaïne de mécanisme inconnu). Après activation, des substances vasoactives, telles que l’histamine, la sérotonine, la tryptase, l’héparine, des cytokines et des prostaglandines, sont libérées et peuvent conduire à des réactions systémiques [1, 3].
Selon la classification de l’Organisation mondiale de la santé (OMS) de 2016, la mastocytose est une entité autonome figurant parmi les néoplasies hématopoïétiques.
Clinique et diagnostic
La mastocytose se manifeste par une prolifération néoplasique clonale de mastocytes anormaux, qui s’agglomèrent dans la peau (mastocytose cutanée) ou, plus rarement, dans la moelle osseuse ou d’autres organes extra-cutanés (mastocytose systémique) [2, 3, 5, 6]. Les mastocytes anormaux présentent différentes formes, des granules cytoplasmiques atténués et répartis inégalement, ainsi que des noyaux cellulaires fusiformes atypiques [2]. La cause de la prolifération néoplasique clonale de mastocytes est dans la plupart des cas une mutation ponctuelle somatique «gain de fonction» du récepteur KIT sur le codon 816 [1–6].
Selon la classification de l’OMS de 2016, le diagnostic de mastocytose systémique peut être posé lorsque le critère majeur et un critère mineur sont remplis ou lorsque trois critères mineurs sont remplis (tab. 1).
Tableau 1: Critère majeur et critères mineurs selon la classification de l’OMS de 2016 (adapté d’après [6]).
Critère majeur
Mise en évidence d’infiltrats multifocaux de mastocytes, c.-à-d. au minimum 15 mastocytes/agrégat, dans la moelle osseuse et/ou d’autres organes extra-cutanés
Critères mineurs
>25% de mastocytes immatures ou atypiques dans le frottis de moelle osseuse ou dans d’autres organes extra-cutanés
Mise en évidence d’une mutation ponctuelle de KIT sur le codon 816 dans les mastocytes de la moelle osseuse, du sang ou d’autres organes extra-cutanés
Expression de CD25 avec ou sans CD2 par les mastocytes de la moelle osseuse, du sang ou d’autres organes extra-cutanés, en plus des marqueurs des mastocytes normaux
Tryptase sérique >20 µg/l de façon persistante
Une tryptase sérique accrue est présente chez pratiquement tous les patients atteints de mastocytose systémique; elle fait office de paramètre de dépistage, mais n’est pas suffisante pour retenir le diagnostic. Par ailleurs, une valeur normale n’exclut pas une mastocytose systémique.
Chez notre patient, le critère majeur ainsi que l’ensemble des quatre critères mineurs étaient remplis.
Les altérations cutanées maculo-papuleuses hyperpigmentées formant de petites taches, dues à des infiltrats de mastocytes dans la peau, sont typiques. En cas d’irritation cutanée mécanique, une rougeur urticarienne (signe de Darier) est observée [3]. Une anomalie cutanée n’est toutefois pas présente chez tous les patients (mastocytose médullaire) [1, 6].
Concernant les mastocytoses systémiques, qui sont surtout diagnostiquées chez les adultes entre 30 et 50 ans, la distinction est faite entre le type indolent, le type «smouldering» et le type agressif. Les hommes et les femmes sont touchés à parts égales. L’espérance de vie des patients atteints de mastocytose systémique indolente n’est pas réduite [1]. Il est très rare que la forme indolente évolue en mastocytose systémique agressive; lorsque c’est le cas, la progression peut être lente ou rapide.
En outre, la combinaison d’une mastocytose systémique et d’une néoplasie hématologique associée peut survenir; la leucémie à mastocytes est plus rare [4]. Une leucémie à mastocytes est présente lorsqu’il y a ≥10% de mastocytes dans le sang périphérique et/ou ≥20% de mastocytes dans l’aspiration médullaire. Une autre variante de la mastocytose est le sarcome à mastocytes [1–6].
Une description détaillée des sous-types de mastocytoses systémiques et des critères diagnostiques correspondants est fournie dans le tableau 2:
Tableau 2: Sous-types de mastocytose systémique (adapté d’après [6]).
Sous-types de mastocytose systémique
Critères diagnostiques Pour tous les sous-types: les critères diagnostiques de la mastocytose systémique sont remplis (selon le tableau 1)
Mastocytose systémique indolente
Aucun des critères C n’est présent (selon le tableau 3)
Pas de mise en évidence d’une néoplasie hématologique associée
Faible charge mastocytaire
Lésions cutanées la plupart du temps présentes
Mastocytose médullaire
Comme décrit plus haut, sauf:
Mise en évidence d’une infiltration de la moelle osseuse
Pas de lésions cutanées
Mastocytose systémique «smouldering»
≥2 critères B, pas de critères C (selon le tableau 3)
Pas de mise en évidence d’une néoplasie hématologique associée
Charge mastocytaire élevée
Mastocytose systémique avec néoplasie hématologique associée
Mise en évidence d’une néoplasie hématologique associée (par ex. syndrome myélodysplasique, néoplasie myéloproliférative, leucémie myéloïde aiguë, lymphomes)
Mastocytose systémique agressive
≥1 critère C (selon le tableau 3)
La plupart du temps, sans lésions cutanées
Leucémie à mastocytes
Biopsie de la moelle osseuse: infiltrats diffus de mastocytes atypiques immatures
Aspiration de moelle osseuse: ≥20% de mastocytes
Forme classique: proportion de mastocytes ≥10% des leucocytes dans le sang périphérique
Plus fréquent: forme aleucémique (proportion de mastocytes <10% des leucocytes dans le sang périphérique)
La plupart du temps, sans lésions cutanées
Contrairement au type indolent, les mastocytoses systémiques de types «smouldering» et agressif se caractérisent par une atteinte d’organes due à l’infiltration de mastocytes, avec ou sans limitation fonctionnelle. La distinction est faite entre les critères B et C. Les critères «B» se rapportent à la souffrance engendrée par la maladie (B pour «burden of disease»). Parmi les critères B, dont au minimum deux doivent être présents pour le sous-type de la mastocytose systémique «smouldering», figurent une charge mastocytaire élevée, des signes de dysplasie ou de myéloprolifération dans les lignées non mastocytaires (mais sans diagnostic définitif de néoplasie hématologique associée), une hépatomégalie sans limitation fonctionnelle, une splénomégalie sans hypersplénisme et une lymphadénopathie. La mastocytose systémique agressive est définie par les critères C. Les critères «C» se rapportent à la nécessité d’un traitement cytoréducteur (C pour «cytoreduction-requiring»). Parmi les critères C figurent une insuffisance médullaire avec au moins une cytopénie, une hépatomégalie avec insuffisance hépatique, une ascite et/ou une hypertension portale, une splénomégalie avec hypersplénisme, une atteinte squelettique avec ostéolyses et une malabsorption avec perte de poids [2, 4, 5, 6]. Les critères B et C sont listés de façon détaillée dans le tableau 3.
Tableau 3: Critères B et C de la mastocytose systémique (B pour «burden of disease» et C pour «cytoreduction-requiring») (adapté d’après [6]).
Critères B
Charge mastocytaire élevée: biopsie de la moelle osseuse avec >30% de mastocytes, tryptase sérique >200 μg/l
Signes de dysplasie ou de myéloprolifération dans les lignées non mastocytaires (mais pas de diagnostic de néoplasie hématologique associée)
Hépatomégalie sans insuffisance hépatique, splénomégalie sans hypersplénisme et/ou lymphadénopathie
Critères C
Dysfonction médullaire induite par l’infiltration de mastocytes avec ≥1 cytopénie: hémoglobine <100 g/l, thrombocytes <100 G/l, granulocytes neutrophiles <1,0 G/l
Hépatomégalie avec insuffisance hépatique, ascite et/ou hypertension portale
Atteinte squelettique avec ostéolyses avec/sans fractures pathologiques
Splénomégalie avec hypersplénisme
Malabsorption avec perte de poids en raison d’une infiltration mastocytaire gastro-intestinale
Les facteurs entrant en ligne de compte en tant que déclencheurs d’une dégranulation des mastocytes incluent les piqûres d’hyménoptères, les changements de température (par ex. lors d’un bain chaud ou d’un saut dans l’eau froide), les infections, le stress émotionnel, les efforts physiques, les irritations mécaniques, les aliments et les boissons tels que le fromage, le chocolat et l’alcool, ainsi que les médicaments tels que les opiacés, les anti-inflammatoires non stéroïdiens, les produits de contraste, les anesthésiques et les relaxants musculaires. Les médiateurs de mastocytes libérés peuvent entraîner prurit, bouffée vasomotrice, urticaire, nausées et vomissements, crampes abdominales, diarrhée, hypotension, syncopes, tachycardie, symptômes neurologiques tels que troubles de la mémoire et de la concentration, céphalées, troubles du sommeil, arthralgies, myalgies et réactions anaphylactiques graves [1, 3, 4].
Il convient de faire la distinction entre la mastocytose systémique et un syndrome d’activation mastocytaire, qui se caractérise par une augmentation réactionnelle du nombre et de l’activité des mastocytes, mais sans multiplication néoplasique clonale des mastocytes. Le syndrome d’activation mastocytaire donne lieu à des symptômes similaires à ceux de la mastocytose systémique, mais les critères diagnostiques de la mastocytose systémique ne sont pas remplis [3].
Traitement de la mastocytose systémique
Il n’existe jusqu’à présent pas de traitement curatif. Le traitement est symptomatique, et cytoréducteur en cas de forme agressive [2]. Les déclencheurs d’une dégranulation mastocytaire mentionnés ci-dessus doivent être évités. Si une utilisation est inévitable, une prémédication par antihistaminiques et glucocorticoïdes est nécessaire. En raison du risque accru de réaction anaphylactique grave, par ex. après une piqûre d’hyménoptère, les patients doivent emporter avec eux un kit d’urgence avec un antihistaminique et un glucocorticoïde, ainsi qu’un auto-injecteur d’adrénaline. La prise quotidienne d’un antihistaminique H1 non sédatif (par ex. cétirizine ou desloratadine) est recommandée pour la prophylaxie des symptômes induits par la dégranulation mastocytaire. En cas de symptômes gastro-intestinaux, un antihistaminique H2 peut en outre être prescrit (par ex. ranitidine). Les glucocorticoïdes (par ex. budésonide) peuvent être utiles en cas de réactions anaphylactiques fréquentes, de diarrhée, de malabsorption et d’ascite [3]. Pour les formes agressives, un traitement cytoréducteur est nécessaire. Toutefois, jusqu’à présent, seules quelques études non randomisées avec de petits nombres de cas ont été conduites à sujet [1]. En tant que traitement de première ligne dans le cadre des évolutions lentement progressives, il est possible d’administrer en off-label un interféron α ou de la cladribine (analogue de la désoxyadénosine). Pour les formes rapidement progressives, la cladribine ou l’inhibiteur de tyrosine kinase midostaurine (Rydapt®, autorisé pour les mastocytoses systémiques avancées) ainsi que des chimiothérapies d’association avec transplantation de cellules souches hématopoïétiques allogéniques sont des options discutées. L’inhibiteur de tyrosine kinase imatinib (Glivec®), qui lie le KIT, n’est pas efficace en cas de mutation ponctuelle p.D816V-KIT, qui est le plus souvent à l’origine de la mastocytose systémique [2–6].
Chez notre patient, sous traitement médicamenteux par antihistaminique H1 à dose élevée (desloratadine 3 × 5 mg/jour), deux nouveaux épisodes avec sensation de malaise et vertiges sont survenus par la suite; les symptômes ont toutefois rapidement régressé après la prise des médicaments de secours décrits ci-dessus. Sous ce traitement médicamenteux, nous évaluons le risque de nouvelle survenue de symptômes de dégranulation mastocytaire systémiques comme étant très faible, raison pour laquelle nous avons soutenu la reprise du travail du patient dans le domaine des transports publics.
Le tableau clinique de la mastocytose systémique est très hétérogène et non spécifique, la liste des diagnostics différentiels est longue, et la pose du diagnostic n’est donc pas facile, comme ce fut le cas chez notre patient. En cas d’anaphylaxie, de syncope, de bouffées vasomotrices, de prurit ou d’altérations cutanées brunes et d’urticaire, ainsi que de troubles gastro-intestinaux d’origine indéterminée et de céphalées, il convient toujours de penser à une mastocytose systémique ou à une hypertryptasémie persistante. La tryptase sérique est considérée comme un paramètre de dépistage. Toutefois, une valeur accrue n’est pas suffisante pour la pose du diagnostic et une valeur normale n’exclut pas une mastocytose systémique. Le diagnostic est confirmé par examen de la moelle osseuse et examen de génétique moléculaire. La réalisation d’une ostéodensitométrie est pertinente suite à la pose du diagnostic. En cas de symptômes gastro-intestinaux avec avant tout une diarrhée, une coloscopie avec prélèvement biopsique doit être conduite. Dans ce cadre, une infiltration mastocytaire du côlon est souvent diagnostiquée à tort comme colite à éosinophiles.
Outre les contrôles cliniques, il convient de réaliser des déterminations régulières de la tryptase sérique et de l’hémogramme différentiel afin d’évaluer l’évolution de la mastocytose systémique indolente et une éventuelle transition vers une forme agressive.
L’essentiel pour la pratique
• En cas d’anaphylaxie, de syncope, de bouffées vasomotrices, de prurit ou d’altérations cutanées brunes et d’urticaire, ainsi que de troubles gastro-intestinaux d’origine indéterminée et de céphalées, il convient toujours de penser à une mastocytose systémique.
• La tryptase sérique est considérée comme un paramètre de dépistage. Toutefois, une valeur accrue n’est pas suffisante pour la pose du diagnostic et une valeur normale n’exclut pas une mastocytose systémique.
• Pour confirmer le diagnostic, un examen de la moelle osseuse et un examen de génétique moléculaire sont nécessaires, et les critères diagnostiques de la classification de l’OMS de 2016 doivent être remplis.
• Le traitement est symptomatique. L’inhibiteur de tyrosine kinase midostaurine représente une nouvelle option thérapeutique pour les mastocytoses systémiques avancées.
• Il est essentiel que le patient soit équipé de médicaments d’urgence et d’un auto-injecteur d’adrénaline, et qu’il fasse l’objet d’une évaluation avant les interventions chirurgicales/dentaires. Il existe un risque accru de réactions graves après les piqûres d’hyménoptères, y compris chez les personnes qui ne sont pas allergiques.
Disclosure statement
Les auteurs n’ont pas déclaré des obligations financières ou personnelles en rapport avec l’article soumis.
Correspondance
Dr méd. Jeroen S. Goede Medizinische Onkologie und Hämatologie Kantonsspital Winterthur Brauerstrasse 15 CH-8401 Winterthur jeroen.goede[at]ksw.ch
Références
1 Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis and related disorders. N Engl J Med. 2015;373(2):163–72.
2 Pardanani A, Tefferi A. Systemic mastocytosis in adults: 2017 update on diagnosis, risk stratification and management. Am J Hematol. 2016;91(11):1146–59.
3 Horny HP, Sotlar K, Valent P, Hartmann K. Die Mastozytose. Dtsch Arztebl. 2008;105(40):686–92.
4 Valent P, Sperr WR, Akin C. How I treat patients with advanced systemic mastocytosis. Blood. 2010;116:5812–17.
5 Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420–27.
6 Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th ed. International Agency for Research on Cancer; 2017.