

Publié le 01.09.2021
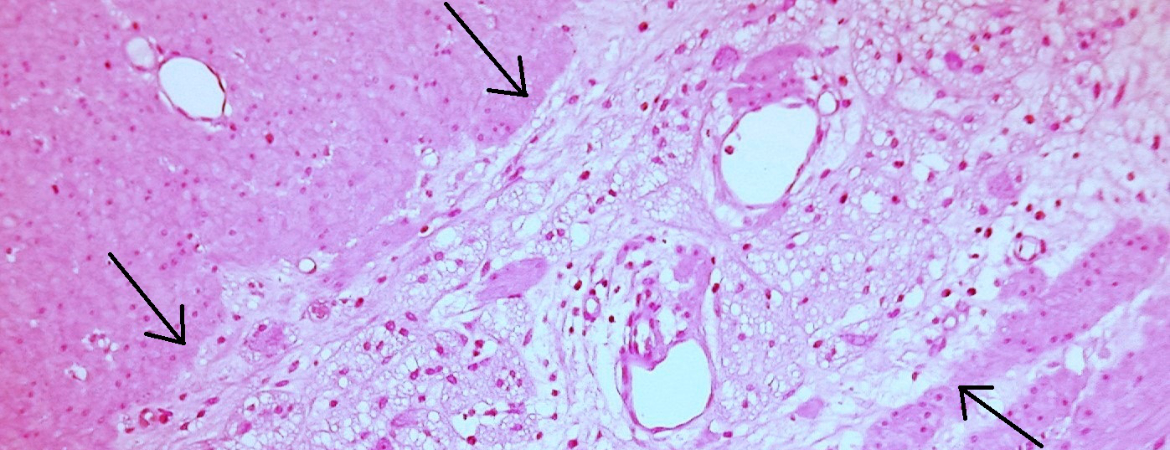
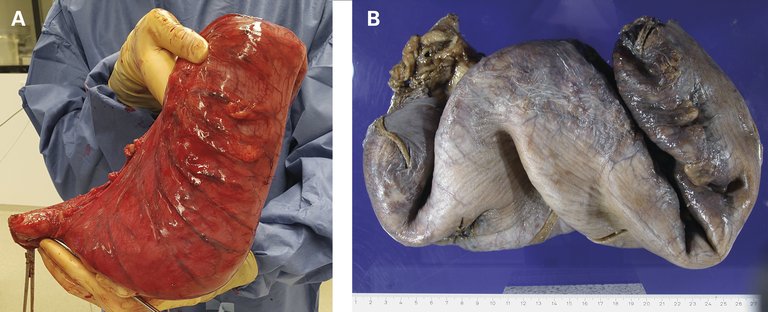
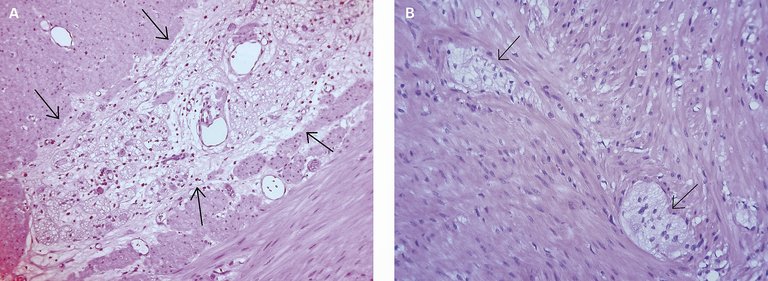
Un jeune patient de 15 ans atteint de constipation chronique s’est présenté à la clinique pour enfant avec un abdomen aigu extrêmement gonflé.


Publié sous la licence du droit d'auteur.
"Attribution - Non-Commercial - NoDerivatives 4.0"
Pas de réutilisation commerciale sans autorisation..
See: emh.ch/en/emh/rights-and-licences/
