


Publié le 07.12.2021
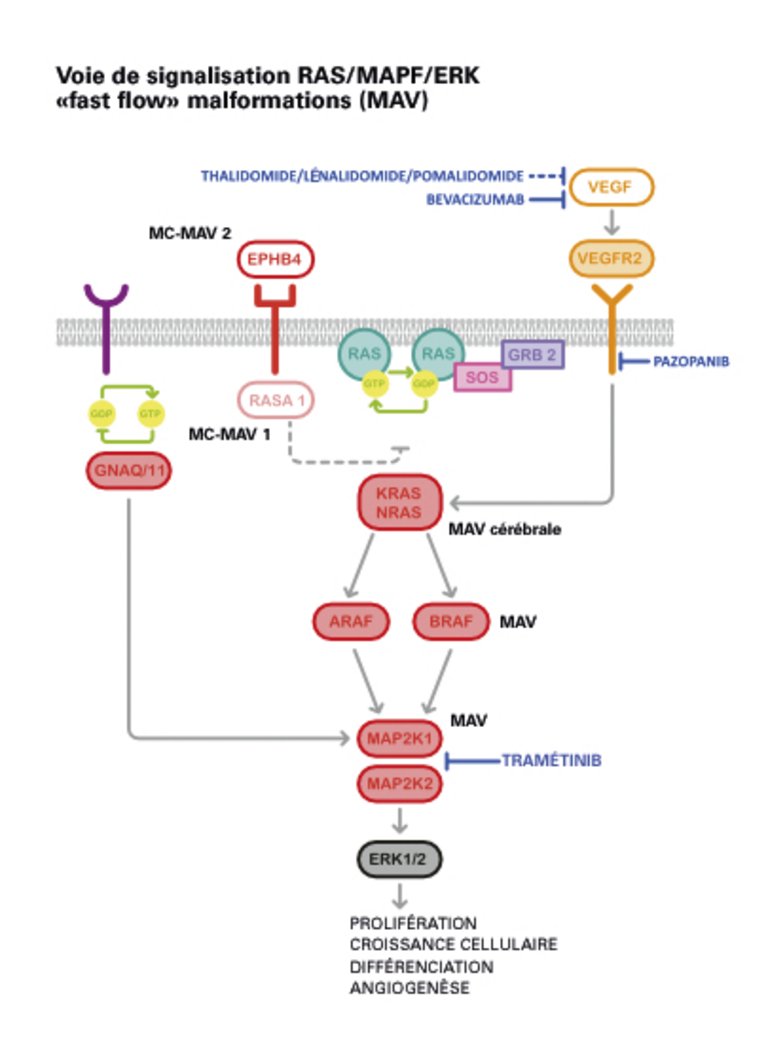
| Tableau 1: Classification des malformations vasculaires congénitales de la Société internationale pour l’étude des anomalies vasculaires (ISSVA) [2]. |
| Malformations vasculaires simples |
| Un type de vaisseau est touché de manière isolée: malformation artérielle (MA), malformation veineuse (MV), malformation lymphatique (ML), malformation capillaire (MC) y compris malformations artérioveineuses (MAV). |
| Malformations vasculaires combinées |
| Au moins deux types de vaisseau sont touchés: diverses combinaisons, p. ex. malformation capillaro-veineuse (MCV), malformation capillaro-veino-lymphatique (MCVL). |
| Malformations tronculaires de vaisseaux embryologiquement matures et médicalement identifiables |
| Le terme «tronculaire» est dérivé de «truncus» et caractérise de grands vaisseaux sanguins ou lymphatiques embryologiquement matures. Parmi les malformations vasculaires tronculaires se trouvent les aplasies, hypoplasies, ectasies et les anévrismes dégénératifs non inflammatoires. |
| Malformations vasculaires associées à d’autres anomalies |
| Diverses malformations vasculaires congénitales sont associées à des anomalies des os, organes viscéraux et parties molles (généralement hypertrophie, rarement hypotrophie) et sont souvent désignées par un syndrome éponyme. Les syndromes de malformations vasculaires les plus fréquents sont: le syndrome de Klippel-Trénaunay (MC + MV +/− ML + hypertrophie des extrémités) et le syndrome de Parkes-Weber (MC + MV +/− ML + MAV + hypertrophie des extrémités). |
| Malformations vasculaires congénitales non classifiées |
| Tableau 2: Symptomatologie clinique des malformations artérioveineuses congénitales périphériques (classification de Schobinger) [3, 4]. | |
| Stade | Tableau clinique |
| I Phase de repos (quiescence) | – Tache cutanée vasculaire et hyperthermie possible – Shunts AV identifiables à l’échographie Doppler – MAV est présente, mais n’entraîne aucun symptôme clinique |
| II Phase évolutive (expansion) | Stade I plus: – Agrandissement – Pulsation et frémissement palpable – Bruit de machine audible – Veines de drainage artérialisées dilatées/tortueuses |
| III Phase destructive | Stade II plus: – Dystrophie cutanée, ulcérations non cicatrisantes (syndrome de vol sous-clavier) – Saignements des ulcérations ou muqueuses cutanées – Nécroses tissulaires et lésions osseuses lytiques possibles |
| IV Phase de décompensation | Stade III plus: – Insuffisance cardiaque avec débit cardiaque accru – Résistance vasculaire périphérique (RVP) anormalement réduite – Hypertension veineuse avec modifications cutanées secondaires (ulcères de la jambe) |
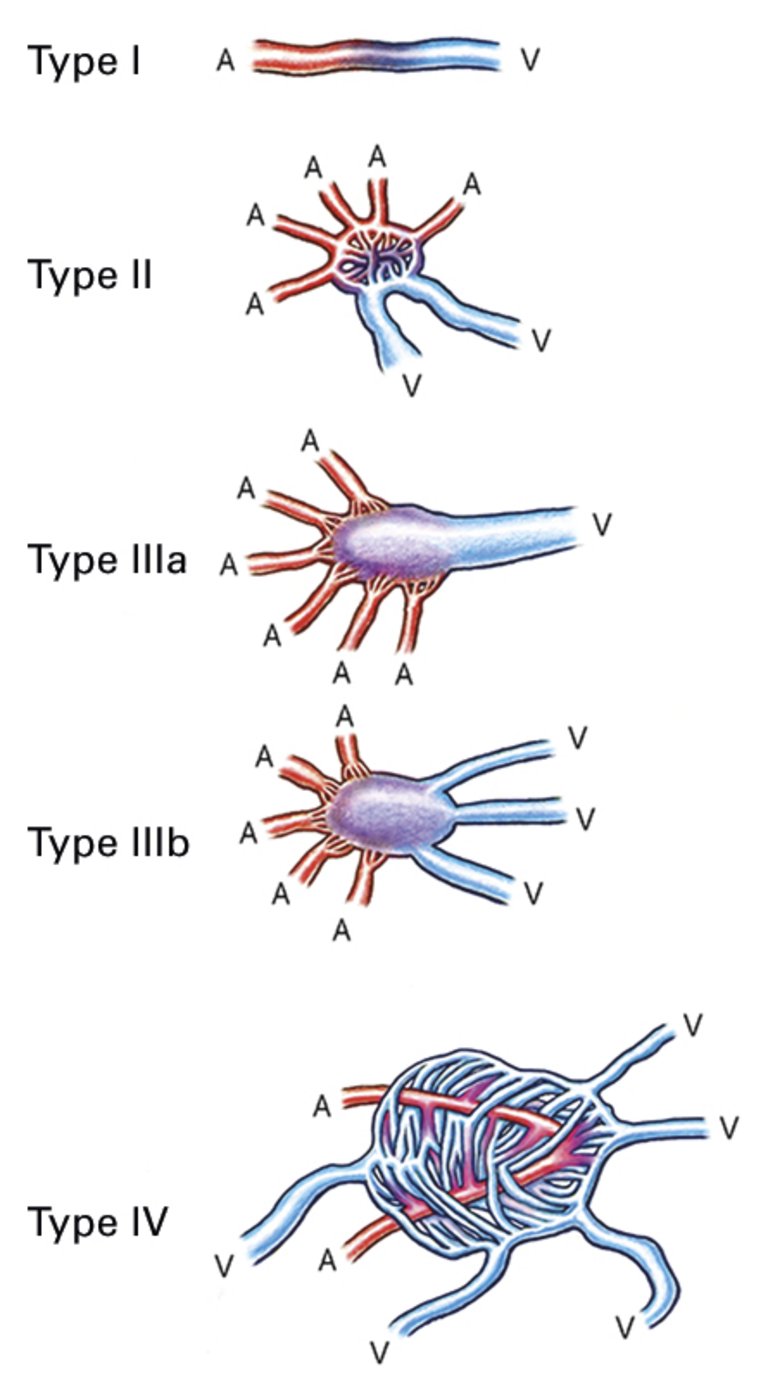
Publié sous la licence du droit d'auteur.
"Attribution - Non-Commercial - NoDerivatives 4.0"
Pas de réutilisation commerciale sans autorisation..
See: emh.ch/en/emh/rights-and-licences/
