Les fièvres récurrentes auto-inflammatoires sont caractérisées par des épisodes fébriles périodiques avec une inflammation systémique et, en fonction de l’affection, spécifique aux organes. L’interleukine-1β y joue un rôle central.
Pathogenèse
Les mécanismes pathogéniques des fièvres récurrentes auto-inflammatoires se déroulent essentiellement dans le système immunitaire inné non adaptatif. Il n’y a pas de formation d’auto-anticorps ou de réponse des cellules T spécifiques à un antigène, comme c’est typiquement le cas pour les maladies auto-immunes et allergiques. Il se produit bien plus souvent une sécrétion accrue d’interleukine (IL-) 1, régulée par l’inflammasome, un complexe multiprotéique cytoplasmique.
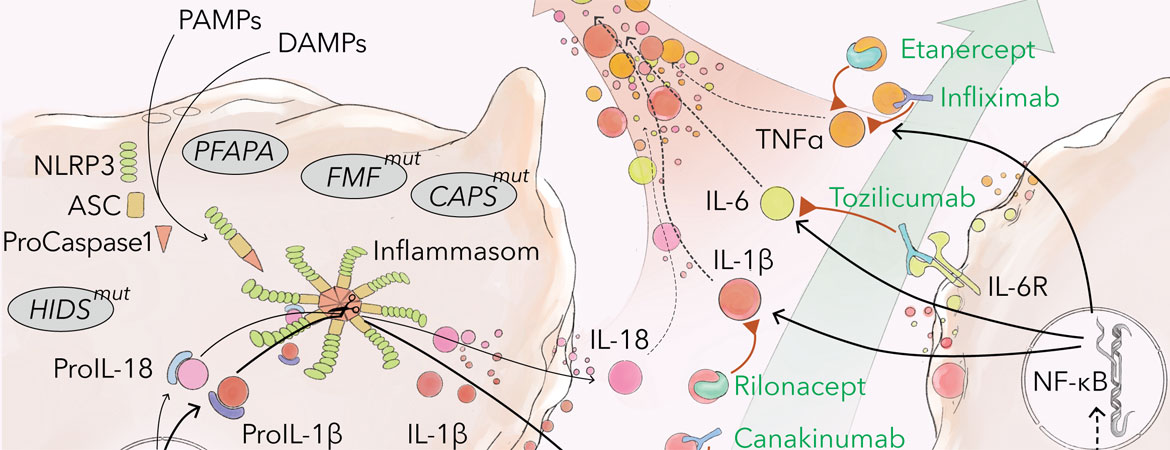
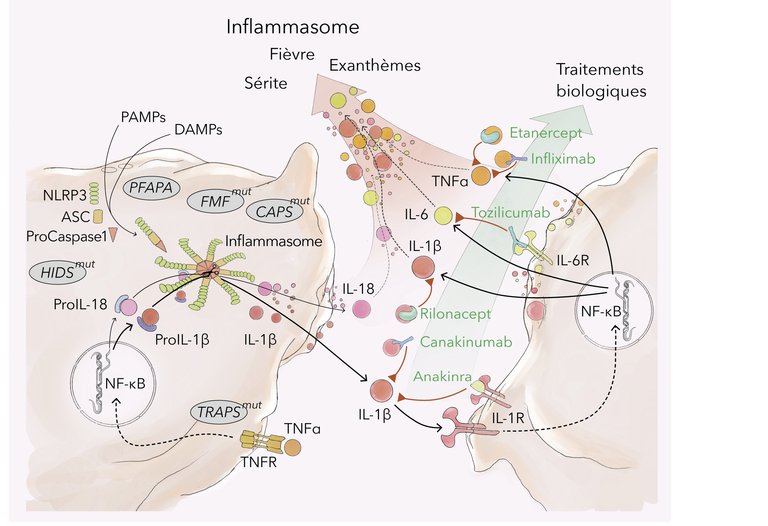
L’inflammasome, un complexe constitué de protéines sensorielles («pattern recognition receptors» [PRRs]), de protéines adaptatrices et de protéases (caspases), est formé et activé par des signaux de danger extrinsèques et intrinsèques (entre autres «pathogen-associated molecular patterns» [PAMPs], tels que lipopolysaccharides [LPS], «damage-associated molecular patterns» [DAMPs], tels qu’adénosine triphosphate [ATP], suite à des dommages cellulaires) [1–3] (cf. fig. 1 à la fin de l’article). Son activation entraîne notamment la conversion de la pro-caspase-1 inactive en caspase-1 active, qui à son tour convertit les précurseurs des cytokines pro-inflammatoires IL-1β et IL-18 dans leur forme active. L’IL-1β est principalement sécrétée par les monocytes/macrophages. Il s’agit d’un médiateur de réactions inflammatoires locales et systémiques avec production de cytokines pro-inflammatoires, qui contrôle ainsi entre autres la fièvre, la tolérance à la douleur, la vasodilatation, l’hypotension, ainsi que l’infiltration de cellules immunitaires dans le tissu endommagé [3–6].
Diagnostic et complications
Il convient de songer à une fièvre récurrente auto-inflammatoire en cas d’épisodes récurrents de fièvre, dont l’évolution n’est toutefois pas progressive, accompagnés d’une élévation des paramètres inflammatoires. L’affection s’accompagne préférentiellement d’une inflammation cutanée, d’une inflammation articulaire, ainsi que de sérites. Des intervalles asymptomatiques séparent souvent les épisodes fébriles.
Une anamnèse approfondie des poussées fébriles est essentielle pour la pose du diagnostic. Il convient d’interroger le patient sur le début, la durée, les symptômes concomitants, l’absence de symptômes entre les épisodes, les déclencheurs potentiels (infections mineures, menstruations chez les femmes, stress, activité physique, froid, fatigue), les traitements pris jusqu’alors et la réponse à ces traitements, les antécédents familiaux et l’origine ethnique. Dans le cadre du diagnostic différentiel, il faut exclure une cause infectieuse (en particulier endocardite), maligne (entre autres lymphomes) et auto-immune (entre autres lupus érythémateux systémique), vascularites, une immunodéficience ou une cause médicamenteuse. L’absence de périodes asymptomatiques entre les épisodes fébriles est par exemple évocatrice d’une cause auto-immune, tandis qu’une évolution progressive avec symptômes B est évocatrice d’une cause maligne. La réponse à une antibiothérapie plaide en faveur d’une cause infectieuse. L’âge lors de la première manifestation et la durée des poussées fébriles peuvent constituer des renseignements utiles dans le cadre des fièvres récurrentes héréditaires (tab. 1) [7, 8].
Tableau 1: Vue d’ensemble des fièvres récurrentes auto-inflammatoires monogéniques.
Critères mineurs: accès incomplet touchant >1 des sites 1.–3., 4. douleurs des membres inférieurs à l’effort, 5. réponse à la colchicine
Critères d’appoint: 1. antécédents familiaux positifs de FMF, 2. origine ethnique compatible, 3. âge <20 ans lors de la première manifestation, 4.–7. caractéristiques des accès: sévères (nécessitant un repos au lit), rémission spontanée, intervalles asymptomatiques, réponse inflammatoire, 8. hématurie/protéinurie épisodique, 9. laparotomie sans particularités, 10. chez des apparentés
FMF: fièvre méditerranéenne familiale; CAPS: syndrome périodique associé à la cryopyrine; FCAS: «familial cold autoinflammatory syndrome»; SMW: syndrome de Muckle-Wells; CINCA/NOMID: «chronic infantile neurologic cutaneous articular syndrome/neonatal onset multisystem inflammatory disease»; TRAPS: syndrome périodique associé au récepteur du facteur de nécrose tumorale; HIDS/MKD: syndrome d’hyper-immunoglobulines D / déficit en mévalonate kinase; AINS: anti-inflammatoires non stéroïdiens.
Il est fréquent que les patients soient asymptomatiques, avec des paramètres inflammatoires normaux, lors d’une consultation ordinaire. Il est dès lors essentiel que les patients se présentent lors d’une poussée afin de procéder à une évaluation clinique, à une objectivation des anomalies, à une détermination des paramètres inflammatoires et à un prélèvement d’hémocultures pour pouvoir exclure une cause infectieuse. En cas de suspicion d’une maladie auto-inflammatoire, le patient devrait être adressé à une clinique spécialisée pour y bénéficier d’examens complémentaires et d’un traitement.
Pour les fièvres récurrentes auto-inflammatoires monogéniques, des tests génétiques sont recommandés en cas de manifestations cliniques concordantes [4, 9]. Le dosage des cytokines ne s’est pour l’instant pas établi dans la pratique pour la pose du diagnostic; la réponse aux inhibiteurs des voies de signalisation de l’IL-1 s’avère plus utile. Les valeurs d’IL-1 peuvent être normales chez les patients atteints d’une maladie auto-inflammatoire [10–12]. Les formes héréditaires sont associées à un risque d’amyloïdose AA et donc de développement d’une néphropathie amyloïde, raison pour laquelle il est indispensable de procéder à des contrôles réguliers de la protéine C réactive, de la sérum amyloïde A et de la fonction rénale, y compris protéinurie, avec évaluation de l’activité de la maladie sous traitement [13, 14].
Fièvres récurrentes auto-inflammatoires
Fièvres récurrentes héréditaires
Les fièvres récurrentes héréditaires correspondent à des altérations d’origine génétique de l’inflammasome ou des protéines impliquées dans sa régulation.
La fièvre méditerranéenne familiale (FMF), qui est une affection monogénique de transmission autosomique récessive, est la fièvre récurrente auto-inflammatoire la plus fréquente à travers le monde. Elle est causée par une mutation du gène «MEditerrean FeVer» (MEFV), avec formation d’un inflammasome pyrine. Il convient de songer à cette affection chez les personnes originaires du bassin méditerranéen/Moyen-Orient qui présentent des poussées fébriles récidivantes, le plus souvent accompagnées de sérites [15]. Un accès typique est défini comme une fièvre récurrente (>38 °C) qui dure de 12 heures à 3 jours. Les accès incomplets sont des épisodes douloureux répétitifs avec entre autres 1–2 des caractéristiques suivantes: température <38 °C, épisodes d’une durée plus ou moins longue, absence de signes de péritonite en cas de douleurs abdominales. Un accès de FMF en est véritablement un uniquement s’il remplit les critères d’un accès typique ou incomplet. Les critères diagnostiques de la FMF de Livneh et al. sont listés dans le tableau 1. Le diagnostic de FMF est posé en présence de ≥1 critère majeur ou ≥2 critères mineurs ou 1 critère mineur plus ≥5 critères d’appoint [16]. La réponse à la colchicine corrobore le diagnostic de FMF [14, 17, 18].
Le syndrome périodique associé à la cryopyrine («cryopyrin associated periodic syndrom», CAPS), qui regroupe l’urticaire familiale au froid («familial cold autoinflammatory syndrome», FCAS), le syndrome de Muckle-Wells (SMW) et le syndrome chronique infantile neurologique cutané articulaire/ maladie inflammatoire multisystémique néonatale («chronic infantile neurological cutaneous articular syndrome/ neonatal onset multisystem inflammatory disease», CINCA/NOMID), est une maladie de transmission autosomique dominante avec des mutations dans le gène NLRP3 («NOD-like receptor 3» ou «cold-induced autoinflammatory syndrome 1» [CIAS1]), qui code pour la protéine cryopyrine. Il se manifeste par des poussées fébriles, des exanthèmes de type urticarien et des arthralgies. Le FCAS est typiquement caractérisé par des poussées auto-limitantes (en l’espace de 24 heures) déclenchées par le froid. Le SMW, en revanche, n’est pas déclenché par le froid et peut être responsable d’une surdité. Le CINCA/NOMID représente la forme la plus grave; il débute au cours des premières semaines de vie, a une évolution chronique et s’accompagne de déformations squelettiques [18, 19].
Le syndrome périodique associé au récepteur du facteur de nécrose tumorale («tumor necrosis factor receptor-associated periodic syndrome», TRAPS) est une maladie de transmission autosomique dominante dont les symptômes s’apparentent à ceux de la FMF. Toutefois, les épisodes fébriles durent plus longtemps (5 jours à plusieurs semaines) et ne s’améliorent pas sous colchicine. Il est causé par des mutations faux-sens (missense) dans le gène TNFRSF1A, qui code pour le récepteur du facteur de nécrose tumorale (TNFR). La maladie survient principalement en Europe centrale et en Europe du Nord. Le syndrome d’hyper-immunoglobulines (Ig) D (HIDS), ou fièvre périodique hollandaise, appelé ainsi suite aux premières descriptions de la maladie en Hollande et du fait des taux sériques élevés d’IgD lors des poussées, est aujourd’hui aussi dénommé «fièvre périodique associée à un déficit en mévalonate kinase» (MKD) sur la base du déficit enzymatique sous-jacent. La maladie est causée par une mutation dans le gène de la mévalonate kinase, une enzyme jouant un rôle clé dans la biosynthèse du cholestérol [17, 18, 20].
Les principales caractéristiques des fièvres récurrentes auto-inflammatoires monogéniques sont résumées dans le tableau 1 [21].
Diagnostics différentiels des poussées fébriles chez l’adulte
PFAPA est l’abréviation de «periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis», ou «fièvre périodique, stomatite aphteuse, pharyngite, adénite cervicale» en français. La cause exacte est pour l’heure inconnue; des variants de mutations dans les gènes associés à l’inflammasome (NLRP3, MEFV) sont discutés. La maladie survient le plus souvent avant l’âge de cinq ans et régresse spontanément durant la puberté. Les critères cliniques de Marshall sont listés dans le tableau 2. Ces critères possèdent une bonne sensibilité, mais une faible spécificité. Un diagnostic différentiel majeur est la neutropénie cyclique. Une amygdalectomie peut conduire à une rémission [22–24].
La maladie de Still est une forme particulière de polyarthrite rhumatoïde. On distingue deux formes: l’une survient chez les enfants (arthrite juvénile idiopathique systémique [AJIS]), tandis que l’autre survient chez les adultes (maladie de Still de l’adulte [MSA]). La cause exacte n’est pour l’instant pas connue, mais un orage cytokinique est néanmoins postulé, le plus souvent précédé d’un déclencheur infectieux. Contrairement à la polyarthrite rhumatoïde, des facteurs rhumatoïdes augmentés ou des anticorps antinucléaires ne peuvent pas être mis en évidence. Le diagnostic est posé sur la base de critères cliniques (tab. 2). A l’aide du profil cytokinique, il est possible de faire la distinction entre le phénotype systémique (IL-1ß, IL-18) et le phénotype articulaire (facteur de nécrose tumorale [TNF-] α, IL-6) [25]. Une complication grave et potentiellement fatale de la maladie de Still est le syndrome d’activation macrophagique (SAM), qui devrait être recherché en cas de cytopénie [26]. Parmi les autres diagnostics différentiels figurent le syndrome de Schnitzler et le syndrome de Sweet. Le syndrome de Schnitzler est caractérisé par un exanthème urticarien non prurigineux et par une gammapathie monoclonale. Le diagnostic est posé sur la base des critères de Strasbourg [27, 28]. Le syndrome de Sweet est une dermatose aiguë neutrophilique. Ses principales caractéristiques incluent fièvre, lésions érythémateuses de type nodules/plaques, numération accrue des neutrophiles, ainsi qu’association avec des tumeurs malignes hématologiques, une polyarthrite rhumatoïde et des maladies inflammatoires chroniques de l’intestin.
Tableau 2: Vue d’ensemble des diagnostics différentiels des poussées fébriles chez l’adulte.
Diagnostics différentiels des poussées fébriles chez l’adulte
Génétique/mutation
Age
Durée des poussées fébriles
Manifestations cliniques
Traitement
Syndrome PFAPA
Inconnue, variants NLRP3/ MEFV postulés dans l’inflammasome
<5 à env. 35 ans
Durée de la fièvre de 3–6 jours, toutes les 3–8 semaines
Critères diagnostiques de Marshall: fièvre récurrente débutant avant la 5e année de vie, avec symptômes constitutionnels sans signes d’infection des voies respiratoires supérieures et ≥1 des critères suivants: 1. stomatite aphteuse, 2. lymphadénite cervicale, 3. pharyngite; exclusion d’une neutropénie cyclique; intervalles asymptomatiques entre les poussées; pas de troubles de la croissance/du développement
Colchicine, anakinra, canakinumab
Maladie de Still de l’adulte (MSA)
Polygénique
Tout âge
Fièvre quotidienne durant plusieurs semaines
Diagnostic: au min. 5 critères dont 2 critères majeurs – Critères majeurs: fièvre >39 °C durant ≥1 semaine, arthralgies/arthrite d’une durée ≥2 semaines, exanthème rose saumon non prurigineux, leucocytose >10 000/mm3 avec granulocytose >80% – Critères mineurs: maux de gorge, lymphadénopathie, splénomégalie, tests de la fonction hépatique anormaux, anticorps antinucléaires et facteurs rhumatoïdes négatifs; exclusion de tumeurs malignes, d’infections, d’une polyarthrite rhumatoïde/de vascularites; typique: valeurs accrues de ferritine; complication: SAM
Episodes fébriles récurrents de quelques jours à quotidiens
Critères de Strasbourg: – 2 critères obligatoires: exanthème urticarien non prurigineux, gammapathie monoclonale – ≥2 critères mineurs: fièvre récurrente (>38 °C), anomalies du remodelage osseux (scintigraphie osseuse, IRM) ou élévation de la phosphatase alcaline osseuse, infiltrats cellulaires riches en neutrophiles à la biopsie cutanée, leucocytose ou élévation de la CRP
Corticoïdes, AINS, anakinra, canakinumab
Syndrome de Sweet (dermatose aiguë fébrile neutrophilique)
Inconnue
30–50 ans, avant tout femmes
Poussées fébriles récurrentes, durée de 5–14 jours, tous les 22–35 jours
Neutropénie (neutrophiles ≤500/mm3), fatigue, pharyngite, ulcères buccaux, stomatite, cellulite, lymphadénopathie, en association avec des tumeurs malignes hématologiques, des maladies inflammatoires chroniques de l’intestin ou une polyarthrite rhumatoïde
Les principaux (d’après nous) diagnostics différentiels (non complets) des poussées fébriles chez l’adulte après l’exclusion d’une origine infectieuse, maligne et auto-immune sont résumés dans le tableau 2.
Possibilités thérapeutiques en cas de fièvres récurrentes (auto-)inflammatoires
Les corticoïdes représentent une option thérapeutique appropriée durant les poussées aiguës du syndrome PFAPA. Une dose unique de corticoïdes (0,5–2 mg/kg de poids corporel [PC]/jour) est administrée au cours d’une poussée et en cas de non-réponse, une deuxième dose est administrée le lendemain. L’administration de corticoïdes peut mettre un terme à la fièvre, mais peut néanmoins entraîner un raccourcissement des intervalles entre les épisodes. Les corticoïdes peuvent également être utiles pour contrôler l’activité de la maladie en cas de CAPS, de HIDS/MKD, de TRAPS, de MSA et de syndrome de Schnitzler. Les administrations prolongées de corticoïdes devraient néanmoins être évitées en raison du vaste profil d’effets indésirables [2].
La colchicine à la dose de 0,5 mg deux fois par jour jusqu’à au maximum 3 mg/jour chez les adultes représente le traitement de premier choix en cas de FMF. Elle est également utilisée en cas de syndrome PFAPA. Dans la FMF, elle agit avant tout en empêchant l’activation de l’inflammasome pyrine [15, 29]. Les effets indésirables incluent entre autres des troubles gastro-intestinaux (diarrhées, nausées, douleurs abdominales) et des leucopénies. Elle est généralement considérée comme bien tolérée et est sûre durant la grossesse. La prise continue de colchicine à vie sert non seulement à prévenir les accès de la maladie, mais réduit également le risque d’amyloïdose [2, 14, 17, 18].
Les inhibiteurs du TNF-α (infliximab, étanercept) sont utilisés en tant que traitement de troisième ligne en cas de MSA [26]. Le profil d’effets indésirables des inhibiteurs du TNF-α englobe un risque accru d’infections par des mycobactéries, des champignons et d’autres agents pathogènes opportunistes [2, 18].
Dans la plupart des maladies auto-inflammatoires, les inhibiteurs de la voie de signalisation de l’IL-1 se sont avérés être les médicaments les plus efficaces. En fait partie l’anakinra (Kineret®), un antagoniste recombinant des récepteurs de l’IL-1, qui est administré en injection sous-cutanée (s.c.) à la dose de 100 mg une fois par jour en raison de sa courte demi-vie. Il est autorisé pour le CAPS et la polyarthrite rhumatoïde (Etats-Unis).
L’inhibiteur de l’IL-1 de plus longue durée d’action rilonacept (Arcalyst®) est une protéine de fusion recombinante constituée des domaines de liaison des parties extracellulaires du récepteur de l’IL-1 et d’une protéine accessoire du récepteur de l’IL-1 liés à la portion Fc de l’immunoglobuline humaine G1 (IgG1). Il est administré une fois par semaine par voie s.c. à la dose de 160 mg et est autorisé pour le CAPS hors NOMID (Etats-Unis).
Le canakinumab est un anticorps IgG1 monoclonal humanisé dirigé contre l’IL-1β. Il est injecté par voie s.c. toutes les huit semaines à la dose de 150 mg [30]. Il est autorisé pour le CAPS, l’AJIS, la MSA, la FMF, le HIDS, le TRAPS, ainsi que la goutte résistante aux traitements (Union Européenne, Etats-Unis). En Suisse, le canakinumab est autorisé par Swissmedic pour les fièvres récurrentes héréditaires et l’AJIS.
Les effets indésirables des inhibiteurs de l’IL-1 incluent un risque accru d’infections virales et bactériennes (infections des voies respiratoires supérieures, urosepsis, méningite) et des réactions locales au site d’injection. Il est recommandé que les patients se fassent vacciner contre la grippe et les pneumocoques avant le traitement; les vaccins vivants sont contre-indiqués durant le traitement [28].
En cas de contrôle insuffisant de la FMF sous colchicine, un traitement par inhibiteurs de l’IL-1 peut être tenté. Dans la MSA, les inhibiteurs de l’IL-1 ont montré une réponse rapide en cas d’atteinte systémique et ont permis une réduction des corticoïdes. Le tocilizumab (Actemra®), un anticorps humanisé dirigé contre le récepteur de l’IL-6, a également montré des résultats prometteurs en cas de MSA systémique et articulaire réfractaire [25, 31]. Dans le syndrome PFAPA et le syndrome de Schnitzler, un blocage de l’IL-1 représente une bonne option en cas de poussées fréquentes.
Les voies de signalisation et les modes d’action des médicaments biologiques sont résumés dans la figure 1.
Figure 1: Voies de signalisation et cibles thérapeutiques en cas de fièvres récurrentes auto-inflammatoires (illustration: Rok Humar, Ayla Yalamanoglu). PAMPs: «pathogen-associated molecular patterns»; DAMPs: «damage-associated molecular patterns»; NLRP3: «NOD-like receptor protein 3»; ASC: «apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain»; NF-kB: «nuclear factor kappa-light-chain-enhancer of activated B-cells»; IL: interleukine; TNF ɑ : facteur de nécrose tumorale alpha; TNFR: récepteur du facteur de nécrose tumorale; HIDS: syndrome d’hyper-immunoglobulines D; FMF: fièvre méditerranéenne familiale; CAPS: syndrome périodique associé à la cryopyrine; PFAPA: fièvre périodique, stomatite aphteuse, pharyngite, adénite cervicale.
L’essentiel pour la pratique
• Les fièvres récurrentes auto-inflammatoires sont rares et elles constituent un diagnostic d’exclusion.
• Elles ont en commun une fièvre périodique et une inflammation, avec souvent une implication de l’inflammasome et une production excessive de cytokines.
• Il est utile que le patient se présente durant une poussée afin de procéder à une détermination des paramètres inflammatoires. En cas de suspicion d’une maladie auto-inflammatoire, le patient devrait être adressé à une clinique spécialisée.
• Le traitement actuel repose en premier lieu sur les corticoïdes, les anti-inflammatoires non stéroïdiens et les médicaments biologiques. Les inhibiteurs de la voie de signalisation de l’interleukine-1 se sont révélés prometteurs pour réduire l’activité de la maladie et les complications, telles que l’amyloïdose AA.
Disclosure statement
Les auteurs ont déclaré ne pas avoir d’obligations financières ou personnelles en rapport avec l’article soumis.
Correspondance
Dr méd. Ayla Yalamanoglu Entzündungssprechstunde Klinik und Poliklinik für Innere Medizin Universitätsspital Zürich Rämistrasse 100 CH-8091 Zürich ayla.yalamanoglu[at]usz.ch
Literatur
1 Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–41.
3 Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol. 2009;124(6):1141–9; quiz 1150–1.
4 Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720–32.
5 Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci. 2019;20(13):3328.
6 Contassot E, Beer HD, French LE. Interleukin-1, inflammasomes, autoinflammation and the skin. Swiss Med Wkly. 2012;142:w13590.
7 Kallinich T, Gattorno M, Grattan CE, de Koning HD, Traidl-Hoffmann C, Feist E, et al. Unexplained recurrent fever: when is autoinflammation the explanation? Allergy. 2013;68(3):285–96.
8 Ahmadinejad Z, Mansouri S, Ziaee V, Aghighi Y, Moradinejad MH, Fereshteh-Mehregan F. Periodic Fever: A Review on Clinical, Management and Guideline for Iranian Patients - Part II. Iran J Pediatr. 2014;24(3):229–40.
9 Shinar Y, Obici L, Aksentijevich I, Bennetts B, Austrup F, Ceccherini I et al. Guidelines for the genetic diagnosis of hereditary recurrent fevers. Ann Rheum Dis. 2012;71(10):1599–605.
10 Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201(9):1479–86.
11 Zhou X, Fragala MS, McElhaney JE, Kuchel GA. Conceptual and methodological issues relevant to cytokine and inflammatory marker measurements in clinical research. Curr Opin Clin Nutr Metab Care. 2010;13(5):541–7.
12 Monastero RN, Pentyala S. Cytokines as Biomarkers and Their Respective Clinical Cutoff Levels. Int J Inflam. 2017;2017:4309485.
13 Lane T, Loeffler JM, Rowczenio DM, Gilbertson JA, Bybee A, Russell TL et al. AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum. 2013;65(4):1116–21.
14 Ozen S, Demirkaya E, Erer B, Livneh A, Ben-Chetrit E, Giancane G, et al. EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis. 2016;75(4):644–51.
15 Fenini G, Contassot E, French LE. Potential of IL-1, IL-18 and Inflammasome Inhibition for the Treatment of Inflammatory Skin Diseases. Front Pharmacol. 2017;8:278.
16 Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40(10):1879–85.
17 Federici S, Caorsi R, Gattorno M. The autoinflammatory diseases. Swiss Med Wkly. 2012;142:w13602.
18 Marino A, Tirelli F, Giani T, Cimaz R. Periodic fever syndromes and the autoinflammatory diseases (AIDs). J Transl Autoimmun. 2019;3:100031.
19 Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, Kone-Paut I, Goldbach-Mansky R, Lachmann H, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017;76(6):942–7.
20 Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis. 2006;1:13.
21 Hansmann S, Lainka E, Horneff G, Holzinger D, Rieber N, Jansson AF, et al. Consensus protocols for the diagnosis and management of the hereditary autoinflammatory syndromes CAPS, TRAPS and MKD/HIDS: a German PRO-KIND initiative. Pediatr Rheumatol Online J. 2020;18(1):17.
22. Vanoni F, Theodoropoulou K, Hofer M. PFAPA syndrome: a review on treatment and outcome. Pediatr Rheumatol Online J. 2016;14(1):38.
23 Kraszewska-Głomba B, Matkowska-Kocjan A, Szenborn L. The Pathogenesis of Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Cervical Adenitis Syndrome: A Review of Current Research. Mediators Inflamm. 2015;2015:563876.
24 Thomas KT, Feder HM Jr, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr. 1999;135(1):15–21.
25 Jamilloux Y, Gerfaud-Valentin M, Henry T, Sève P. Treatment of adult-onset Still’s disease: a review. Ther Clin Risk Manag. 2014;11:33–43.
26 Feist E, Mitrovic S, Fautrel B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat Rev Rheumatol. 2018;14(10):603–18.
27 de Koning HD, Bodar EJ, van der Meer JW, Simon A; Schnitzler Syndrome Study Group. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum. 2007;37(3):137–48.
28 Bonnekoh H, Krause K. Das Schnitzler-Syndrom – eine seltene Differenzialdiagnose und interdisziplinäre Herausforderung. Aktuelle Rheumatologie. 2017;42:53–58.
29 Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol. 2016;17(8):914–21.
30 Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu Rev Med. 2014;65:223–44.
31 De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N Engl J Med. 2018;378(20):1908–19.