Un patient de 43 ans, autrement sain, souffre depuis 1½ ans d’épisodes récidivants de faiblesse générale et d’intolérance à la performance avec hypotension, tachycardie, œdèmes généralisés, vertiges et douleurs musculaires et articulaires diffuses.
Contexte
Nous décrivons le cas d’un patient présentant des symptômes et résultats typiques d’un syndrome de fuite capillaire idiopathique (syndrome de Clarkson). La mise en évidence d’une gammapathie monoclonale, qui est typiquement associée à ce syndrome rare et peu connu, a livré la clé du diagnostic après une longue épreuve douloureuse pour le patient. Sous traitement intraveineux par gammaglobulines hautement dosées, la maladie est en rémission depuis trois ans.
Rapport de cas
Anamnèse
Un patient âgé de 43 ans, autrement sain, souffre depuis un an et demi d’épisodes récidivants de faiblesse générale et d’intolérance à la performance, associées à une hypotension, une tachycardie, des œdèmes généralisés, des vertiges ainsi que des douleurs musculaires et articulaires diffuses. Ces épisodes durent 48–72 heures et se répètent tous les 5–6 jours. Ils s’accompagnent d’une baisse de la fréquence mictionnelle suivie d’une phase polyurique. Durant les épisodes, le patient n’est pas en mesure d’assurer ses tâches professionnelles. Les symptômes le poussent à renoncer à son activité sportive. Il est soumis à des examens cardiologiques et neurologiques, y compris une électromyographie, sans résultat anormal. L’analyse sanguine révèle une légère anémie avec un taux d’hémoglobine de 118 g/l. En termes de diagnostic secondaire, des bandes monoclonales sont détectées dans le liquide cérébrospinal, comme expression de la faible paraprotéinémie systémique de type IgG lambda (non quantifiable) diagnostiquée ensuite par immunofixation du sérum. Le patient est donc soumis à un examen hématologique.
Examen clinique et résultats
Lors de la consultation initiale, le patient est exempt de symptômes, normotendu et normocarde, son poids est de 75 kg pour une taille de 182 cm. Une légère anémie normochrome normocytaire est confirmée avec une valeur d’hémoglobine de 128 g/l et un taux d’hématocrite de 37,2%. Le taux d’albumine s’élève à 42 g/l.
Dix jours plus tard, un examen de la moelle osseuse est réalité pour approfondir le diagnostic de la gammapathie monoclonale de signification indéterminée (MGUS) et la légère anémie. A ce moment, le patient se sent très affaibli, il souffre de vertiges, douleurs musculaires diffuses et œdèmes périphériques. Son poids est monté à 79 kg, il présente une tachycardie sinusale pour une pression artérielle normale. Le taux d’hémoglobine s’élève à 194 g/l, celui d’hématocrite à 56,9%.
Les résultats de l’examen de la moelle osseuse sont normaux, il n’est notamment détecté aucune infiltration due à un myélome multiple ou un lymphome non hodgkinien. L’immunophénotypage ne montre aucune population de cellules plasmatiques clonales. En présence d’une cellularité et morphologie normales de l’hématopoïèse, il n’existe aucune indication de syndrome myéloprolifératif. La recherche de mutation de JAK2 est négative. La paraprotéinémie de type IgG lambda est confirmée, le ratio des chaînes légères libres kappa/lambda est normal. Il n’existe aucune albuminurie. Le CT natif «low dose» du corps entier est normal.
Par la suite, il a été possible de mettre en évidence à plusieurs reprises une hémoconcentration avec hausse de la valeur d’hématocrite de 36 à 62% et du taux d’hémoglobine de 128 à 214 g/l en présence d’autres épisodes de faiblesse, ainsi qu’une hypertension et une tachycardie. La hausse du taux d’hématocrite est liée à une prise de poids allant jusqu’à 5 kg et à une baisse du taux d’albumine jusqu’à 30 g/l (cf. tab. 1 pour les valeurs de laboratoire).
Tableau 1: Valeurs de laboratoire lors de la consultation initiale et pendant un épisode de fuite capillaire.
Valeur
Consultation initiale
Episode
Hémoglobine (g/l)
128
214
Hématocrite (%)
37,2
61,8
Leucocytes (G/l)
5,06
16,28
Neutrophiles (G/l)
3,46
12,98
Albumine (g/l)
42
30
Créatinine (mmol/l)
80
170
Diagnostic
Au vu des épisodes récidivants avec hypertension, hausse du taux d’hématocrite, œdèmes périphériques et hypoalbuminémie sans albuminurie et de la mise en évidence d’une paraprotéine, nous établissons le diagnostic d’un syndrome de fuite capillaire idiopathique (syndrome de Clarkson) associé à une gammapathie monoclonale.
Traitement et évolution
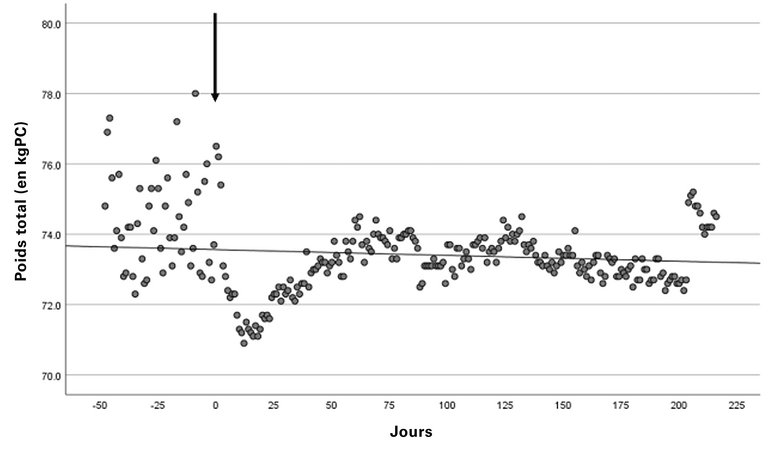
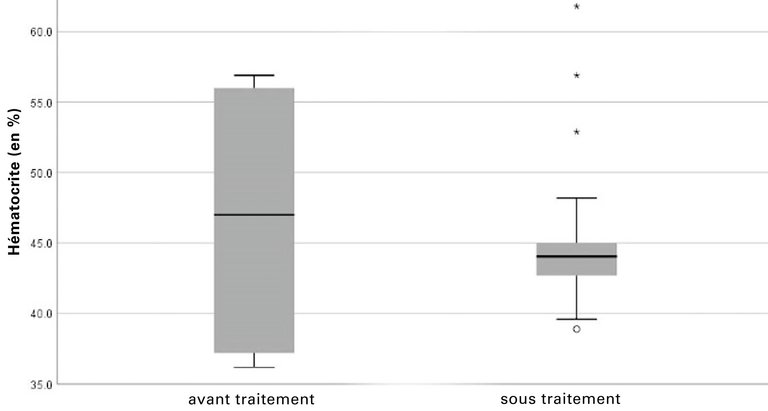
Conformément aux recommandations du «EurêClark Study Group», nous initions un traitement intraveineux par gammaglobulines hautement dosées (2 mg/kg de poids corporel toutes les 4 semaines). Sous ce traitement, les épisodes de fuite capillaire cessent complètement, les symptômes et les fluctuations de poids disparaissent (fig. 1), tout comme la hausse du taux d’hématocrite (fig. 2) et la baisse du taux d’albumine.
Figure 1: Mesure du poids du patient 49 jours avant le début du traitement et jusqu’à 216 jours après. La flèche marque le début du traitement. Il apparaît une nette réduction du poids corporel (avant traitement vs traitement: 74,36 kg ± 1,39 kg vs 73,22 kg ± 0,85 kg; valeur moyenne ± SD). kg: kilogramme; PC: poids corporelle.
Figure 2: Taux d’hématocrite avant et pendant le traitement (46,7% ± 10,2 vs 45,3% ± 5,4; valeur moyenne ± SD).
Depuis le début du traitement, le patient ne sollicite plus aucun arrêt travail et retrouve sa pleine capacité de performance physique. Au bout d’un an, la tentative de réduction de la dose d’immunoglobulines à 1 g/kg de poids corporel toutes les 4 semaines entraîne un nouvel épisode de fuite capillaire, d’où la poursuite du traitement avec 2 g/kg. De même, la prolongation de l’intervalle entre les doses a pour conséquence une récidive. Trois ans après le début du traitement, le patient est toujours en rémission.
Discussion
Le syndrome de fuite capillaire idiopathique a été décrit pour la première fois par B. Clarkson en 1960 [1]. Il a publié le cas d’une femme âgée de 34 ans qui «souffrait d’une perte subite de plasma au niveau du lit vasculaire et chez laquelle se sont développés une hypovolémie et un choc». La maladie était caractérisée par des épisodes répétés de perméabilité capillaire accrue avec œdèmes, hémoconcentration, hypoalbuminémie et hypotension, et une protéine anormale a été mise en évidence dans la fraction des gammaglobulines. La patiente est finalement décédée d’un choc hypovolémique.
En 2010, une série de 25 patients de la «Mayo Clinic», atteints de fuite capillaire idiopathique, a été publiée [2]. Le diagnostic se basait sur une documentation univoque de crises répétées d’hypotension, concentration accrue d’hématocrite, œdèmes périphériques et hypoalbuminémie sans albuminurie. La gammapathie monoclonale n’était pas considérée comme un critère obligatoire, mais mise en évidence chez 19 des 25 patients. La plupart des patients présentaient des symptômes prodromiques tels qu’une fatigue et des vertiges, parfois aussi des myalgies et maux de gorge ainsi qu’une réduction de la fréquence mictionnelle. Durant les épisodes de fuite capillaire est survenue une hausse moyenne du taux d’hématocrite de 19,8% (14,8–24) à 60,5% (53,6–65,6). Le taux moyen d’albumine a baissé de 21 g/l (18–25) pendant un épisode et le taux moyen de créatine a augmenté de 132 µmol/l (88–200). 36% des patients ont développé une rhabdomyolyse, 20% un syndrome des loges qui a parfois nécessité une fasciotomie. La concentration de paraprotéine était typiquement faible (en moyenne 6 g/l). Le taux de progression vers un myélome multiple était de 0,7% par personne-année et ainsi comparable à celui d’une MGUS.
Le «European Clarkson Disease (EurêClark) Registry» est un groupe d’étude international qui collecte les données de patients atteints de syndrome de fuite capillaire idiopathique associé à une gammapathie monoclonale. En 2017, le groupe a publié dans le American Journal of Medicine les résultats thérapeutiques d’une cohorte de 69 patients après administration régulière d’immunoglobuline intraveineuse en prévention des récidives. Les critères pour le diagnostic d’un syndrome de Clarkson étaient: 1. présence d’une gammapathie monoclonale; 2. un ou plusieurs épisodes d’hypovolémie aiguë et œdèmes interstitiels; 3. hémoconcentration avec hypoprotéinémie paradoxale; 4. exclusion d’une autre cause de syndrome de fuite capillaire secondaire ou d’hypoprotéinémie.
La cohorte était composée de 35 femmes et 34 hommes, l’âge moyen était de 52 ans (±12) lors de la première manifestation et 53,5 ans (±12) au moment du diagnostic. Tous les patients avaient une gammapathie de type IgG avec une concentration moyenne de paraprotéine de 4,4 g/l (2–8).
Pour prévenir les épisodes de fuite capillaire, les patients avaient reçu, avant l’an 2000, de la théophylline et de la terbutaline, l’immunoglobuline intraveineuse a ensuite été de plus en plus utilisée comme traitement de première ligne.
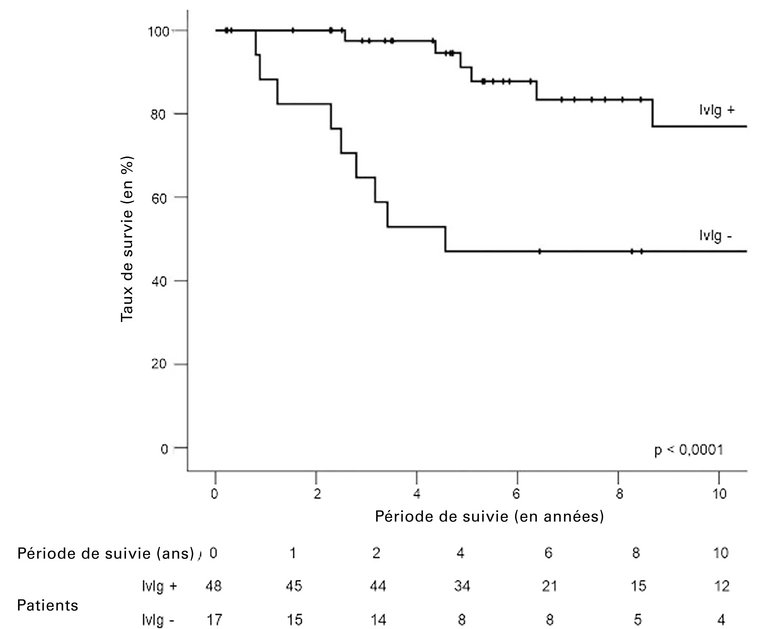
Au cours d’une période moyenne de suivi de 5,1 ans (2,5–9,7), cinq patients ont développé un myélome multiple. 24 sont décédés après une durée moyenne de 3,3 ans (0,9–8): 20 à la suite d’un épisode sévère, quatre d’un myélome multiple. Les patients traités par immunoglobuline intraveineuse ont présenté nettement moins d’épisodes récidivants graves que ceux n’ayant pas reçus d’immunoglobuline intraveineuse (45,8 vs 94,1%) et la survie à 5 et 10 ans était significativement meilleure (91 vs 47% et 77 vs 37%) (fig. 3 [3]).
La pathogenèse du syndrome de fuite capillaire idiopathique et la signification étiologique de la paraprotéine demeurent indéterminées. Des facteurs humoraux entraînant un dysfonctionnement réversible de la barrière des cellules endothéliales microvasculaires sont probablement responsables. Durant les épisodes, des valeurs accrues de facteurs de perméabilité angiogéniques tels que le facteur de croissance de l’endothélium vasculaire (VEGF) et l’angiopoïétine 2 ainsi que d’interleukines et d’autres facteurs inflammatoires ont ainsi été détectées. Aucune modification vasculaire structurelle n’a pu être mise en évidence [4].
Le diagnostic d’un syndrome de fuite capillaire idiopathique reste un diagnostic clinique avec des épisodes récidivants d’hypovolémie aiguë et œdèmes interstitiels, hémoconcentration, hypoalbuminémie sans albuminurie, mise en évidence typique d’une paraprotéine et exclusion d’une autre cause telle que sepsis, anaphylaxie ou angioœdème. Les épisodes sont auto-limitants, durent quelques jours et varient fortement en fréquence et intensité. Un épisode sévère peut entraîner un état de choc potentiellement mortel avec insuffisance rénale aiguë et défaillance multiviscérale. Il peut survenir des thromboses et embolies pulmonaires comme conséquences de l’hémoconcentration, un épanchement péricardique avec tamponnade péricardique et syndrome des loges au niveau des extrémités, ainsi qu’une rhabdomyolyse due aux œdèmes périphériques.
Du point de vue thérapeutique, un traitement de soutien est en premier lieu nécessaire durant la phase aiguë, avec correction de l’hypovolémie par perfusion de solutions cristalloïdes et éventuellement substitution d’albumine. En présence d’évolutions graves, un accompagnement de médecine intensive et l’utilisation de médicaments vasoactifs peuvent être requis, ainsi qu’une anticoagulation en cas de complications thrombotiques. La phase de récupération de la fuite capillaire présente le risque d’une surcharge liquidienne avec œdème pulmonaire, et un traitement diurétique intensif peut être nécessaire. Des cas ont été décrits dans lesquels des saignées ont été effectuées à tort dans l’hypothèse d’une polycythemia vera [5].
Pour éviter de nouveaux épisodes de fuite capillaire, l’utilisation d’immunoglobuline hautement dosée, à une posologie initiale de 2 g/kg de poids corporel toutes les quatre semaines, est recommandée au vu de l’analyse du «European Clarkson Disease (EurêClark) Registry», mentionnée ci-dessus. Cela semble améliorer considérablement l’évolution et le pronostic de la maladie. Aucune donnée d’étude randomisée n’est disponible sur ce traitement et, en raison de la rareté de la maladie, une telle étude n’apparaît pas réalisable. Le mécanisme d’action du traitement par immunoglobulines intraveineuses est incertain. Le traitement est supposé présenter des effets immunomodulateurs et anti-idiotypiques. Il est toutefois non spécifique et onéreux.
L’essentiel pour la pratique
• Le syndrome de fuite capillaire idiopathique est une maladie rare et peu connue présentant des épisodes récidivants et potentiellement mortels d’hyperperméabilité capillaire qui entraîne hypotension, hypoalbuminémie, œdèmes interstitiels ainsi qu’une hausse du taux d’hématocrite due à une hémoconcentration.
• Le syndrome est typiquement associé à une gammapathie monoclonale de signification indéterminée (MGUS).
• En raison de la rareté de la maladie et des symptômes non spécifiques, le diagnostic n’est souvent établi que longtemps après la survenue des premiers symptômes et la maladie est éventuellement sous-diagnostiquée.
• Sur le plan thérapeutique, l’hypotension/hypovolémie et l’insuffisance circulatoire qui en résulte doivent être traitées durant la phase aiguë.
• La prévention des récidives implique un traitement par immunoglobulines intraveineuses hautement dosées.
Disclosure statement
Les auteurs ont déclaré ne pas avoir d’obligations financières ou personnelles en rapport avec l’article soumis.
1 Clarkson B, Thompson D, Horwith M, Luckey EH. Cyclical edema and shock due to increased capillary permeability. Am J Med. 1960;29:193–216.
2 Kapoor P, Greipp PT, Schaefer EW, Mandrekar SJ, Kamal AH, Gonzalez-Paz NC, et al. Idiopathic Systemic Capillary Leak Syndrome (Clarkson’s Disease): The Mayo Clinic Experience. Mayo Clin Proc. 2010;85(10):905–12.
3 Pineton de Chambrun M, Gousseff M, Mauhin W, Lega JC, Lambert M, Rivière S, Dossier A, et al. Intravenous Immunoglobulins Improve Survival in Monoclonal Gammopathy-Associated Systemic Capillary-Leak Syndrome. Am J Med. 2017;130(10):1219.e19–1219.e27.
4 Druey KM, Greipp PR. Narrative Review: The Systemic Capillary Leak Syndrome. Ann Intern Med. 2010;153(2):90.
5 Doubek M, Brychtova Y, Tomiska M, Mayer J. Idiopathic systemic capillary leak syndrome misdiagnosed and treated as polycythemia vera. Acta hamatol. 2005; 113(2):150–1.