La présentation autonome en urgence d’une patiente âgée de 31 ans a eu lieu en raison de taches brunâtres sur le visage, accompagnées d’œdèmes récidivants présents depuis trois mois au niveau du visage et du cou.
Description du cas
La présentation autonome en urgence d’une patiente âgée de 31 ans originaire d’Angola a eu lieu en raison de taches brunâtres sur le visage, accompagnées d’œdèmes récidivants présents depuis trois mois au niveau du visage et du cou. La patiente a par ailleurs rapporté des démangeaisons sur la nuque et une sensation de brûlure du cuir chevelu sous la douche. Outre les manifestations cutanées, elle a fait état d’une perte de force et de douleurs lors du travail impliquant les bras. De plus, elle présentait une sensation de lourdeur et des douleurs dans le bas des jambes, progressives depuis deux ans selon l’anamnèse.
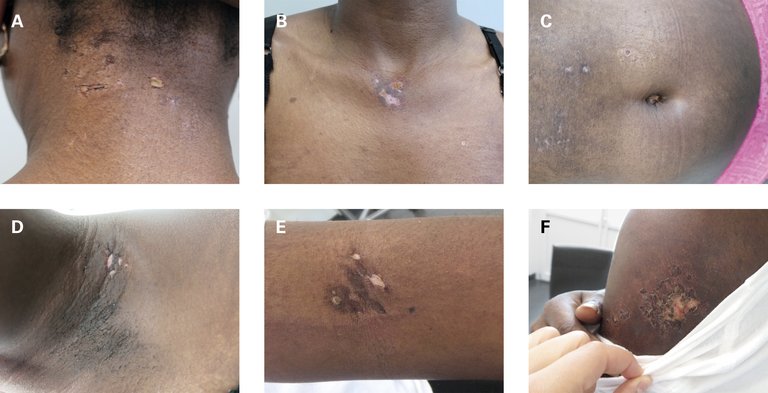
L’examen clinique a révélé une patiente afébrile dans un bon état général, avec un type de peau VI selon Fitzpatrick, une xérose cutanée, des macules hyperpigmentées sur le visage et de multiples petites cicatrices survenues après grattage selon l’anamnèse, ainsi que de multiples érosions d’aspect estampé, généralement petites et nettement délimitées dans la région inguinale, nucale, présternale, abdominale, sur les bras et le cou (fig. 1). Les mains présentaient une hyperkératose de la bordure proximale de l’ongle (toutefois état après manicure). L’examen clinique n’a pu déterminer aucune faiblesse musculaire.
Figure 1: Symptômes cutanés lors la présentation initiale: nucal (A), jugulaire (B), abdominal (C), axillaire droit (D), cubital gauche (E), au bras gauche (F). Un consentement éclairé écrit est disponible pour la publication.
Question 1: Quels sont les diagnostics différentiels auxquels il faut penser?
a) Excoriations par grattage et ulcérations en présence de prurit
b) Consommation de cocaïne
c) Collagénose
d) Cause infectieuse
e) a–d
Initialement, des excoriations par grattage ont été suspectées car la patiente a rapporté un prurit prononcé. Le dépistage du prurit réalisé dans un premier temps (détermination de l’hémogramme, créatinine, protéine C-réactive [CRP], ferritine, valeurs hépatiques, thyréostimuline [TSH], hémoglobine A1c [HbA1c], sérologie du virus de l’immunodéficience humaine [VIH], test de détection d’helminthes) n’a toutefois fourni aucun résultat concluant. Une consommation de cocaïne a été discutée comme cause exogène possible, car celle-ci est associée à des lésions pseudo-vasculitiques. La cocaïne coupée au lévamisole (anthelminthique utilisé en médecine vétérinaire) montre en particulier une association avec une vascularite ou vasculopathie cutanée et peut provoquer les ulcérations présentées à la figure 1 [1]. Interrogée, la patiente a rapporté une consommation occasionnelle de cannabis, mais nié l’utilisation de cocaïne. Nous avons ensuite pensé au diagnostic différentiel d’une cause eczémateuse sous forme de dermatose prurigineuse. L’anamnèse ne présentait néanmoins aucune indication d’asthme, de pollinose, ni de dermatite atopique et le taux d’immunoglobulines E (IgE) totales ainsi que la tryptase affichaient des valeurs normales. De même, les analyses infectiologiques (concernant la syphilis, la tuberculose cutanée, la trichophytie profonde, la leishmaniose, etc.) se sont toutes révélées normales. Il n’y avait aucun indication de maladie bulleuse auto-immune en présence d’immunofluorescence normale sans signe de dépôts d’anticorps ou de complément et d’anticorps de pemphigoïde bulleuse ou de pemphigus non accrus à la sérologie. La biopsie cutanée initiale a fourni un résultat histologique non spécifique sans indication de lupus érythémateux ni de dermatomyosite. Le tableau clinique ne présentait aucune modification cutanée typique, telle que des papules de Gottron, un érythème héliotrope et un signe en V ou du châle, suggérant une dermatomyosite. Toutefois, étant donné que des évolutions ulcéreuses sont aussi décrites en cas de dermatomyosite et que, outre les douleurs musculaires et la perte de force symétrique, les analyses biochimiques indiquaient un taux accru de créatine kinase (CK) de 557 U/l (valeur normale <145 U/l), la suspicion de dermatomyosite s’est confirmée. De manière compatible, une hausse des anticorps antinucléaires (ANA) de 1 : 160 a été mise en évidence, le taux de CK a continué d’augmenter jusqu’à 2 818 U/ml (en plus d’une hausse des transaminases et de la lactate déshydrogénase [LDH]).
Question 2: Quelles prochaines étapes diagnostiques sont alors envisageables?
a) Détermination des auto-anticorps associés à la myosite
b) Imagerie par résonance magnétique des muscles concernés
c) Biopsie des muscles concernés
d) Electromyographie (EMG)
e) Tous les examens mentionnés
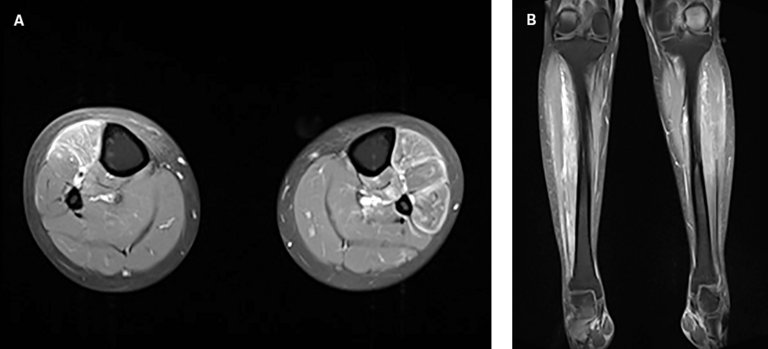
En présence de douleurs musculaires progressives et d’un taux de CK croissant, un examen électrophysiologique a ensuite été réalisé, révélant des potentiels de faible amplitude dans le muscle tibial antérieur et des signes discrets d’une activité spontanée de la musculature paravertébrale au niveau Th12 comme indication possible d’une myosite. L’électromyographie peut être utile pour faire la distinction entre une pathologie musculaire et une faiblesse musculaire d’origine neuropathique. Des modifications caractéristiques peuvent corroborer le diagnostic suspecté d’une dermatomyosite, mais ne sont pas univoques sur le plan diagnostique, car des modifications similaires sont observées en cas de myosites infectieuses, toxiques ou métaboliques [2]. L’imagerie par résonance magnétique (IRM) de la partie inférieure des jambes réalisée en complément a révélé un œdème musculaire bilatéral étendu avec absorption pathologique du produit de contraste, indiquant une myosite bilatérale aiguë (fig. 2).
Figure 2: Imagerie par résonance magnétique de la partie inférieure de la jambe au niveau frontal (A) et transversal (B); séquence T1 avec produit de contraste (en blanc). Œdème musculaire bilatéral étendu avec absorption pathologique du produit de contraste.
La tomodensitométrie ne permet pas de faire la distinction entre une rhabdomyolyse, une dystrophie musculaire et une myopathie métabolique. L’imagerie permet toutefois une biopsie ciblée dans la zone musculaire de l’inflammation et réduit le risque d’une erreur d’échantillonnage [3]. L’examen histologique de la biopsie musculaire a révélé des agrégats plaquettaires dans le périmysium et l’endomysium, mais pas de fibres musculaires atrophiques. Cela correspond à un résultat fortement non spécifique qui, avec le tableau clinique de la patiente, est compatible avec une dermatomyosite, toutefois sans caractère probant. Une biopsie musculaire n’est pas nécessaire chez toutes les personnes concernées lorsque le diagnostic peut déjà être établi au vu de l’examen clinique et des résultats de laboratoire [2]. En conjonction avec la sérologie de myosite par western blot positive pour les anticorps anti-«transcription intermediary factor»-(TIF)1-γ, une dermatomyosite a finalement pu être diagnostiqué chez notre patiente.
Question 3: Quelle est la suite de la procédure la plus indiquée?
a) Poursuite du diagnostic, traitement uniquement après obtention des résultats
b) Protection contre les rayonnements UV, traitement local
c) Traitement systémique
d) Traitement local et systémique
e) Protection contre les rayonnements UV, traitement local et systémique, poursuite du diagnostic
La dermatomyosite est une myopathie inflammatoire idiopathique accompagnée d’une dermatite, qui est cliniquement hétérogène et peut être difficile à diagnostiquer. Les manifestations cutanées (signe du châle, érythème héliotrope, œdème du visage, papules de Gottron, signe du holster, signe de flagellés) varient et peuvent survenir en même temps que la myosite ou de manière décalée (précédemment dans jusqu’à 30% des cas). Les adultes atteints de dermatomyosite ont un risque accru de présence ou d’apparition d’une pathologie maligne. L’établissement du diagnostic de tumeur maligne peut alors avoir lieu avant, pendant ou après la pose du diagnostic de la dermatomyosite – il se fait le plus souvent en même temps ou durant la première année après le diagnostic de dermatomyosite. En plus de l’initiation du traitement, d’autres étapes diagnostiques à la recherche d’une tumeur sont donc urgemment indiquées pour identifier une potentielle néoplasie. Les évolutions ulcéreuses de la dermatomyosite, comme dans le cas de notre patiente, semblent être plus souvent associées à une cause paranéoplasique ainsi qu’à des anticorps anti-TIF1-γ ou anti-«melanoma differentiation-associated protein 5» (MDA5) au western blot.
Sur le plan thérapeutique, nous avons mis en place un traitement systémique de la dermatomyosite en plus d’un traitement anti-inflammatoire local par glucocorticoïdes (classe III) et d’un traitement antiprurigineux systémique par des antihistaminiques.
Question 4: Quelle déclaration concernant la dermatomyosite anti-TIF1-γ-positive est fausse?
a) Le risque de maladie maligne est fortement accru par rapport à la dermatomyosite antiTIF1γnégative.
b) Le plus souvent, il existe une association avec le cancer ovarien, le cancer mammaire ou le lymphome non hodgkinien.
c) Un dépistage annuel de cancer est recommandé pendant au moins 3 à 5 ans après établissement du diagnostic de dermatomyosite.
d) Les personnes anti-TIF1-γ-positives présentent plus souvent une pneumopathie interstitielle concomitante, un phénomène de Raynaud ou une arthrite.
e) Les personnes souffrant de dermatomyosite anti-TIF1-γ-positive présentent une atteinte cutanée plus étendue.
Le spectre des auto-anticorps permet, dans une certaine mesure, d’émettre un pronostic. Parmi les auto-anticorps spécifiques de la myosite, les anticorps anti-TIF1-γ sont associés à un risque fortement accru de malignité [4, 5]. Jusqu’à 75% des personnes concernées de plus de 40 ans présentent une dermatomyosite associée à une tumeur maligne [6]. Le plus souvent, il existe une association avec les cancers ovariens, cancers mammaires et lymphomes non hodgkiniens, d’où la recommandation d’un dépistage annuel (ou semestriel) pendant au moins 3 à 5 ans après établissement du diagnostic. Les malades présentant des auto-anticorps anti-TIF1-γ ont des symptômes cutanés plus étendus par rapport à ceux exempts d’anti-TIF1-γ, tandis que les pneumopathies interstitielles concomitantes, le phénomène de Raynaud et l’arthrite sont en revanche plus rares [7]. Après traitement réussi de la tumeur maligne, des rémissions cliniques de la dermatomyosite et des anticorps anti-TIF1-γ sérologiquement régressifs voire indétectables peuvent être observés [4, 8]. Une nouvelle hausse des anticorps peut indiquer une récidive tumorale ou une augmentation de l’activité de la dermatomyosite [9].
Chez notre patiente, nous avons réalisé un dépistage tumoral détaillé. La tomodensitométrie n’a révélé aucun point de départ cervical, thoracique, abdominal et cérébral d’une tumeur. L’échographie transvaginale n’a fourni aucune indication de cancer ovarien, ni d’autre tumeur gynécologique maligne et le frottis de Papanicolaou (PAP) était normal. De même, les examens diagnostiques approfondis au moyen de la gastroscopie, coloscopie, cytoscopie et fibroscopie oto-rhino-laryngologique (ORL) n’ont fourni aucune indication de pathologie maligne. L’IRM n’a pas non plus montré de suspicion de malignité. La mammographie et l’échographie du tissu mammaire ont toutes deux suscité une suspicion de fibroadénomes bilatéraux. Etant donné que, en particulier en cas de dermatomyosite avec anticorps anti-TIF1-γ, le risque d’une paranéoplasie est présent et que les carcinomes mammaires ne sont pas rares, plusieurs biopsies mammaires ont été réalisées malgré un résultat radiologique plutôt bénin, apportant la confirmation histologique des fibroadénomes. Aucune pathologie sous-jacente maligne n’a ainsi pu être diagnostiquée jusqu’à présent chez la patiente.
Question 5: Quel est le traitement systémique de première ligne en cas de dermatomyosite?
a) Méthotrexate ou azathioprine
b) Hydroxychloroquine
c) Glucocorticoïdes
d) Immunoglobulines
e) Rituximab
Malgré les divers effets indésirables à long terme, les glucocorticoïdes systémiques sont utilisés en traitement de première ligne. Nous avons mis en place un traitement stéroïdien systémique hautement dosé par méthylprednisolone en intraveineuse pendant trois jours, suivie de prednisone par voie orale (1 mg/kg).L’œdème du visage ainsi que les douleurs musculaires ont alors régressé. Par ailleurs, il est apparu une amélioration positive des modifications cutanées avec réépithélialisation totale des ulcérations et une baisse du taux de CK à l’analyse biochimique. Afin d’obtenir un traitement épargnant le plus possible les stéroïdes, celui-ci a été complété par l’azathioprine (100 mg/jour). Le traitement concomitant par des immunosuppresseurs, tels que le méthotrexate, l’azathioprine, le mofétil mycophénolate ou le cyclophosphamide, permet de réduire la dose de stéroïdes pour l’induction de la rémission ainsi que le risque de récidive pendant le sevrage des glucocorticoïdes [10]. Etant donné que l’effet de l’azathioprine n’est attendu qu’au bout de deux à six mois et que notre patiente continuait de présenter une forte activité pathologique, le traitement a été complété par l’administration d’immunoglobulines (2 g / kg de poids corporel toutes les 4–8 semaines). Les immunoglobulines intraveineuses peuvent être administrées en complément en cas de maladie grave ou d’évolution réfractaire, sont efficaces chez 70% des personnes concernées et s’accompagnent d’une amélioration de la force musculaire, souvent observée dès la première perfusion. Toutefois, l’effet est d’une durée limitée et des perfusions répétées sont nécessaires [11, 12]. Le rituximab présente une évidence restreinte en termes d’efficacité pour la dermatomyosite cutanée. Il est en particulier employé en cas de myosite réfractaire ou de pneumopathie associée à une dermatomyosite. Concernant les médicaments antipaludiques tels que l’hydroxychloroquine, des effets positifs ont été observés sur les manifestations cutanées, mais pas sur la myosite [12]. Deux ans après l’établissement du diagnostic, notre patiente est, sous prednisone (2,5 mg/jour) et administration régulière d’immunoglobulines intraveineuses, actuellement en rémission durable et toujours exempte de tumeur.
Discussion
Sur peux foncées, les maladies dermatologiques peuvent présenter un tableau clinique nettement différent de celui des peaux claires et les modifications érythémateuses sont justement plus difficiles à reconnaître. Nous n’avons donc pas envisagé en premier lieu une dermatomyosite comme cause des ulcérations. L’établissement du diagnostic n’a pas été facile chez cette patiente et a duré au total plusieurs mois. Outre de nombreuses consultations ambulatoires chez différents dermatologues, plusieurs présentations dans divers services d’urgence ont préalablement eu lieu. Le diagnostic a été rendu difficile, d’une part, par les symptômes cutanés non caractéristiques de la dermatomyosite et, d’autre part, par les résultats histologiques non spécifiques des biopsies cutanées sans indication d’une dermatite de l’interface associée à une collagénose. L’anamnèse présentant des douleurs et une faiblesse musculaires pour un taux de CK initialement seulement légèrement accru nous a ouvert la voie et mené à poursuivre les étapes diagnostiques. Au cours des examens suivants, que ce soit l’imagerie par résonance magnétique, les analyses électrophysiologiques ou histologiques, des modifications non pathognomoniques toujours non spécifiques ont étés observées. La biopsie musculaire n’a permis de mettre en évidence aucune fibre musculaire atrophique, les modifications histologiques étaient non spécifiques et équivoques pour le diagnostic. Ce n’est qu’en réunissant tous les outils diagnostiques actuellement disponibles, en particulier les anticorps spécifiques de la myosite, que le diagnostic a enfin pu être établi. Notre manque d’expérience médicale avec les dermatoses sur peaux foncées a peut-être été un autre facteur ayant contribué à retarder le diagnostic. Dans une étude, 47% du personnel médical de dermatologie était d’avis que la formation ne suffit pas pour diagnostiquer les maladie cutanées sur peaux foncées, ce qui est notamment dû à la sous-représentation des symptômes sur peaux noires dans le matériel didactique [13]. Ce manque est progressivement corrigé: outre les manuels spécialisés dans les peux foncées, de plus en plus de sources en ligne présentant des images de dermatoses sur divers degrés de pigmentation sont disponibles.
Réponses:
Question 1: e. Question 2: e. Question 3: e. Question 4: d. Question 5: c.
Sources en ligne pour les dermatoses sur peaux noires
Question 1: e. Question 2: e. Question 3: e. Question 4: d. Question 5: c.
Disclosure statement
Les auteurs ont déclaré ne pas avoir de conflits d’intérêts potentiels.
Correspondance
Eva Akermann Klinik für Allgemeine Innere Medizin/Hausarztmedizin Rorschacher Strasse 95 CH-9007 St. Gallen eva.akermann[at]hin.ch
Références
1 Marquez J, Aguirre L, Muñoz C, Echeverri A, Restrepo M, Pinto LF. Cocaine-Levamisole-induced vasculitis/vasculopathy syndrome. Curr Rheumatol Rep. 2017;19(6):36.
2 Yang SH, Chang C, Lian ZX. Polymyositis and dermatomyositis – challenges in diagnosis and management. J Transl Autoimmun. 2019;2:100018.
3 Del Grande F, Carrino JA, Del Grande M, Mammen AL, Christopher Stine L. Magnetic resonance imaging of inflammatory myopathies. Top Magn Reson Imaging. 2011;22(2):39–43.
4 Dani L, Holmqvist M, Martínez MA, Trallero-Araguas E, Dastmalchi M, Svensson J, et al. Anti-transcriptional intermediary factor 1 gamma antibodies in cancer-associated myositis: a longitudinal study. Clin Exp Rheumatol. 2020;38(1):67–73.
5 De Vooght J, Vulsteke JB, De Haes P, Bossuyt X, Lories R, De Langhe E. Anti-TIF1-γ autoantibodies: warning lights of a tumour autoantigen. Rheumatology (Oxford). 2020;59(3):469–77.
6 Oldroyd A, Sergeant JC, New P, McHugh NJ, Betteridge Z, Lamb JA, et al. The temporal relationship between cancer and adult onset anti-transcriptional intermediary factor 1 antibody-positive dermatomyositis. Rheumatology (Oxford). 2019;58(4):650–5.
7 Alenzi FM. Myositis specific autoantibodies: A clinical perspective. Open Access Rheumatol. 2020;12:9–14.
8 Shimizu K, Kobayashi T, Kano M, Hamaguchi Y, Takehara K, Matsushita T. Anti-transcriptional intermediary factor 1-γ antibody as a biomarker in patients with dermatomyositis. J Dermatol. 2020;47(1):64–8.
9 Ikeda N, Yamaguchi Y, Kanaoka M, Ototake Y, Akita A, Watanabe T, Aihara M. Clinical significance of serum levels of anti-transcriptional intermediary factor 1-γ antibody in patients with dermatomyositis. J Dermatol. 2020;47(5):490–6.
10 Sasaki H, Kohsaka H. Current diagnosis and treatment of polymyositis and dermatomyositis. Mod Rheumatol. 2018;28(6):913–21.
11 Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971–82.
12 Cobos GA, Femia A, Vleugels RA. Dermatomyositis: An update on diagnosis and treatment. Am J Clin Dermatol. 2020;21(3):339–53.
13 Lester JC, Taylor SC, Chren MM. Under-representation of skin of colour in dermatology images: not just an educational issue. Br J Dermatol. 2019;180(6):1521–2.