


Publié le 24.05.2022
Un choc est une défaillance circulatoire avec un approvisionnement insuffisant en oxygène des organes. La partie 1 de cet articel de revue aborde les causes, la physiopathologie et la clinique des quatre formes de choc.
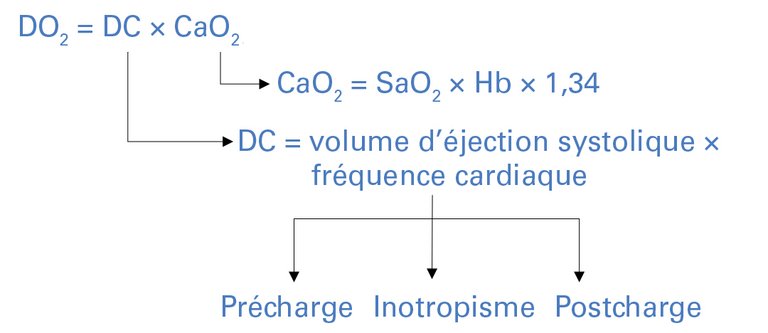
| Tableau 1: Aperçu des quatre formes de choc. | |
| Formes de choc | Mécanismes et causes typiques |
| Choc hypovolémique | Hémorragique: – interne (hémorragie rétropéritonéale) – externe (hémorragie gastro-intestinale, traumatisme) |
| Non hémorragique: – Vomissements, diarrhée | |
| Choc obstructif | Circulation pulmonaire entravée: – Embolie pulmonaire |
| Remplissage cardiaque entravé: – Tamponnade péricardique – Pneumothorax sous tension | |
| Retour veineux vers le cœur entravé: – Syndrome du compartiment abdominal | |
| Choc cardiogénique | Inotropisme perturbé: – Infarctus du myocarde (7–10% de tous les infarctus* [4]) – Myocardite |
| Valvulopathies, défauts anatomiques: – Insuffisance mitrale aiguë dans le cadre d’une rupture du muscle papillaire – Communication interventriculaire | |
| Arythmies: – Tachyarythmies – Bradycardie – Dyssynchronie entre l’oreillette et le ventricule | |
| Choc distributif (inflammatoire) | Infection/ sepsis |
| Polytraumatisme | |
| Opérations majeures | |
| Pancréatite | |
| Anaphylaxie | |
| * Taux de chocs cardiogéniques chez les patientes et patients victimes d’un infarctus du myocarde avec sus-décalage du segment ST (STEMI). | |
| Tableau 2: Diagnostics différentiels de l’hyperlactatémie. |
| Métabolisme anaérobie en cas d’hypoxie tissulaire |
| Choc |
| Effort physique intense, y compris crise d’épilepsie de type «grand mal» |
| Médicaments/intoxications |
| β-agonistes |
| Alcool |
| Metformine |
| Médicaments antirétroviraux |
| Paracétamol |
| Dysfonction hépatique |
| Hyperglycémie, acidocétose diabétique |
| Leucémies et lymphomes |
| Tableau 3: Classification en stades de l’insuffisance rénale aiguë (KDIGO 2012 [15]). | ||
| Stade | Créatinine sérique | Excrétion urinaire |
| 1 | Augmentation de 1,5–1,9 fois ou Augmentation ≥26,5 µmol/l | <0,5 ml/kg/h sur 6–12 h |
| 2 | Augmentation de 2–3 fois | <0,5 ml/kg/h sur ≥12 h |
| 3 | Augmentation de ≥3 fois ou Augmentation ≥354 µmol/l ou Début d’une thérapie de substitution rénale | <0,3 ml/kg/h sur ≥24 h ou Anurie sur ≥12 h |
| KIDGO: «Kidney Disease: Improving Global Outcomes»; h: heure (n). | ||
| Tableau 4: Signes de choc [13]. | |
| Entité clinique | Paramètres |
| Remplissage des vaisseaux | Paramètres vitaux: – Pression artérielle systolique (PAS) <95 mm Hg – Pression artérielle moyenne (PAM) <65 mm Hg – Fréquence cardiaque >100/min |
| Hypoperfusion tissulaire | Peau: – Périphérie froide – Peau marbrée – Temps de remplissage capillaire allongé |
| Saturation en oxygène veineuse centrale (ScvO2) <65% | |
| Taux de lactate dans le sang artériel >2,2 mmol/l | |
| Dysfonctionnements organiques | Encéphalopathie: somnolence, agitation, confusion |
| Insuffisance rénale aiguë: excrétion urinaire <0,5 ml/kg/h | |
| Dysfonction hépatique: augmentation de l’alanine aminotransférase (ALT), de l’aspartate aminotransférase (AST) et de la bilirubine | |
Publié sous la licence du droit d'auteur.
"Attribution - Non-Commercial - NoDerivatives 4.0"
Pas de réutilisation commerciale sans autorisation..
See: emh.ch/en/emh/rights-and-licences/
