PD Dr. med. Christian Rothermundt,
Dr. med. Isabelle Binet,
Prof. Dr. med. Jan Borovicka,
Prof. Dr. med. Oliver Bozinov,
Dr. med. Thomas Clerici,
PD Dr. med. Daniel S. Engeler,
Dr. med. Jeanette Greiner,
Dr. med. Claudia Hader,
Prof. Dr. med. Dr. phil. Karl Heinimann,
Dr. med. Silvia Azzarello-Burri,
Dr. med. Corina Lang,
Dr. med. Ina Krull,
Prof. Dr. med. Sandro J. Stöckli,
PD Dr. med. Aurelius Omlin,
PD Dr. med. Thomas Hundsberger
Kantonsspital St. Gallen, St. Gallen: a Klinik für Medizinische Onkologie und Hämatologie, b Klinik für Nephrologie und Transplantationsmedizin, c Klinik für Gastroenterologie und Hepatologie, d Klinik für Neurochirurgie, e Klinik für Allgemein-, Viszeral-, Endokrin- und Transplantationschirurgie, f Klinik für Urologie, g Klinik für Radiologie und Nuklearmedizin, h Institut für Pathologie, i Augenklinik, j Klinik für Endokrinologie, Diabetologie, Osteologie und Stoffwechselerkrankungen, k Hals-Nasen-Ohrenklinik, l Klinik für Neurologie; m Hämatologie & Onkologie, Ostschweizer Kinderspital St. Gallen, St. Gallen; n Institut für Medizinische Genetik und Pathologie, Universitätsspital Basel, Basel
La maladie de von Hippel-Lindau est un syndrome de predisposition tumorale rare a transmission autosomique dominante. Un depistage standardise doit permettre de detecter les tumeurs au stade asymptomatique et de les traiter precocement.
Introduction
La maladie de von Hippel-Lindau (VHL) est une maladie multisystémique à transmission autosomique dominante, qui est associée à la survenue de multiples tumeurs et kystes, le plus souvent bénins, mais aussi de tumeurs malignes (tab. 1).
Tableau 1: Quand faut-il penser à la maladie de von Hippel-Lindau (VHL)?
Manifestation clinique
Quand penser à la maladie de VHL?
Age typique de survenue [9]
Fréquence en cas de maladie de VHL [9]
Carcinome rénal et kystes rénaux
Carcinomes rénaux bilatéraux et/ou multifocaux Premier diagnostic à un âge ≤45 ans ≥2 membres de la famille atteints de carcinome rénal Autres manifestations typiques de la maladie de VHL
25–50 ans
25–60%
Hémangioblastomes
Hémangioblastomes cérébraux et spinaux
18–35 ans
10–72% (en fonction de la localisation)
Hémangioblastomes rétiniens
12–25 ans
25–60%
Tumeurs du sac endolymphatique
Perte auditive, acouphènes, vertiges
12–45 ans
10–25%
Lésions pancréatiques
En cas de tumeurs neuroendocrines du pancréas et/ou de kystes pancréatiques multiples
24–35 ans
35–70%
Phéochromocytomes et paragangliomes
Avant tout en association avec d’autres manifestations typiques de la maladie de VHL et/ou une anamnèse familiale positive
12–25 ans
10–20%
Cystadénomes épididymaires
Tumeurs bénignes de l’épididyme, souvent bilatérales
14–40 ans
25–60% des hommes
Cystadénomes du ligament large
Tumeurs bénignes du ligament large
16–46 ans
Env. 10% des femmes
La prédisposition est causée par des variants pathogènes de la lignée germinale du gène VHL. L’incidence est de 1:36 000 nouveau-nés et la prévalence est de 1–9/100 000 en Europe [1, 2]. La maladie de VHL présente une pénétrance élevée, avec la survenue de symptômes cliniques chez plus de 90% des personnes touchées jusqu’à l’âge de 65 ans. Les premières tumeurs se développent souvent à l’adolescence. En raison du mode de transmission autosomique dominant, les descendants ont une probabilité de 50% d’hériter du variant VHL pathogène du parent atteint. Chez environ 20% des personnes touchées, le variant VHL pathogène est apparu de novo. L’identification précoce des porteuses et porteurs de la prédisposition à la maladie de VHL et le suivi étroit et régulier par une équipe interdisciplinaire expérimentée sont décisifs pour la réduction de la morbidité spécifique à la maladie, le maintien de la qualité de vie et le pronostic [3].
Le gène VHL, très conservé au cours de l’évolution, fait partie des gènes suppresseurs de tumeurs et il se situe sur le bras court du chromosome 3 (3p25.3). La protéine VHL (pVHL) est impliquée dans de nombreuses voies de signalisation et se compose de deux isoformes, qui possèdent toutes deux une activité de suppression tumorale. La pVHL favorise entre autres la dégradation des «hypoxia-inducible factors» (HIF), qui jouent un rôle majeur dans l’apparition des différentes lésions associées à la maladie de VHL.
La maladie de VHL est souvent diagnostiquée sur la base de critères cliniques, par exemple en présence d’une anamnèse familiale positive et d’une ou plusieurs lésions typiques de la maladie de VHL (deux ou plusieurs lésions du système nerveux central [SNC] ou une lésion du SNC associée à une manifestation viscérale). Même en cas de survenue de lésions typiques isolées, il faut au moins penser à une maladie de VHL. Avant de procéder à un test génétique, il convient d’effectuer un conseil génétique qui, outre les aspects médicaux, aborde également les aspects psychosociaux pour les patientes et patients ainsi que pour leurs proches. La mise en évidence d’un variant pathogène de la lignée germinale du gène VHL permet par la suite aux membres de la famille (en bonne santé) de bénéficier d’un conseil génétique et d’un test génétique (tab. 2) [4].
Tableau 2: Critères pour le conseil et le test génétiques
Indication pour le conseil/test génétiques
Anamnèse familiale négative et deux manifestations associées à la maladie de VHL
Negative Familienanamnese und zwei VHL-assoziierte Manifestationen
Manifestations de la maladie de VHL Angiomes rétiniens Hémangioblastomes cérébraux ou spinaux Tumeurs du sac endolymphatique Carcinomes rénaux Phéochromocytomes, kystes pancréatiques et/ou tumeurs neuroendocrines du pancréas Cystadénomes épididymaires
Présence d’un carcinome rénal et d’au moins un des critères suivants: Premier diagnostic à un âge ≤45 ans Carcinomes rénaux bilatéraux ou multifocaux Anamnèse familiale positive avec ≥2 membres de la famille atteints de carcinome rénal
VHL: von Hippel-Lindau.
Le pronostic de la maladie dépend en grande partie des manifestations concrètes et de la progression des tumeurs.
Manifestations de la maladie de VHL
Les lésions associées à la maladie de VHL se situent principalement dans le SNC, dans la rétine et dans les organes viscéraux.
Des hémangioblastomes du SNC surviennent chez une grande partie des porteuses et porteurs de la caractéristique génétique et sont souvent le premier site de manifestation de la maladie. Ils se situent préférentiellement au niveau du cervelet, du tronc cérébral et de la moelle épinière. Les hémangioblastomes du SNC sont des tumeurs richement vascularisées, souvent accompagnées de la formation de kystes. Des examens neurologiques réguliers ainsi que des examens d’imagerie par résonance magnétique (IRM) cérébrale et spinale sont indispensables pour déterminer le moment optimal pour une intervention neurochirurgicale ou radio-oncologique. En cas d’effet expansif important dans la fosse crânienne postérieure, des situations potentiellement fatales peuvent rapidement apparaître. Les hémangioblastomes du tronc cérébral font partie des lésions les plus difficiles à opérer en raison de leur forte tendance aux hémorragies intra-opératoires et de la fonction essentielle de cette région du cerveau. En plus d’un centre de traitement expérimenté, une réunion de concertation pluridisciplinaire (RCP) dédié à la maladie de VHL est obligatoire afin d’assurer une prise de décision multidisciplinaire et de discuter des alternatives thérapeutiques.
Les hémangiomes capillaires rétiniens sont fréquents, mais peuvent rester longtemps asymptomatiques. Les exsudations rétiniennes, les décollements de rétine, ainsi que les hémorragies vitréennes ou rétiniennes peuvent entraîner une baisse de l’acuité visuelle. Dans la mesure où un traitement (au laser) précoce des lésions est associé à un meilleur pronostic, des examens ophtalmologiques de dépistage réguliers sont indispensables.
Les tumeurs du sac endolymphatique (TSEL), qui sont plutôt rares dans l’ensemble, sont souvent asymptomatiques. En cas d’augmentation de volume, elles entraînent une perte auditive, qui progresse en général lentement. Les acouphènes, les vertiges et la sensation de pression dans l’oreille peuvent en être des symptômes précoces. Les TSEL ne métastasent généralement pas, mais présentent localement une croissance destructrice et infiltrante. Pour les tumeurs à extension limitée, une ablation chirurgicale précoce peut prévenir la perte auditive.
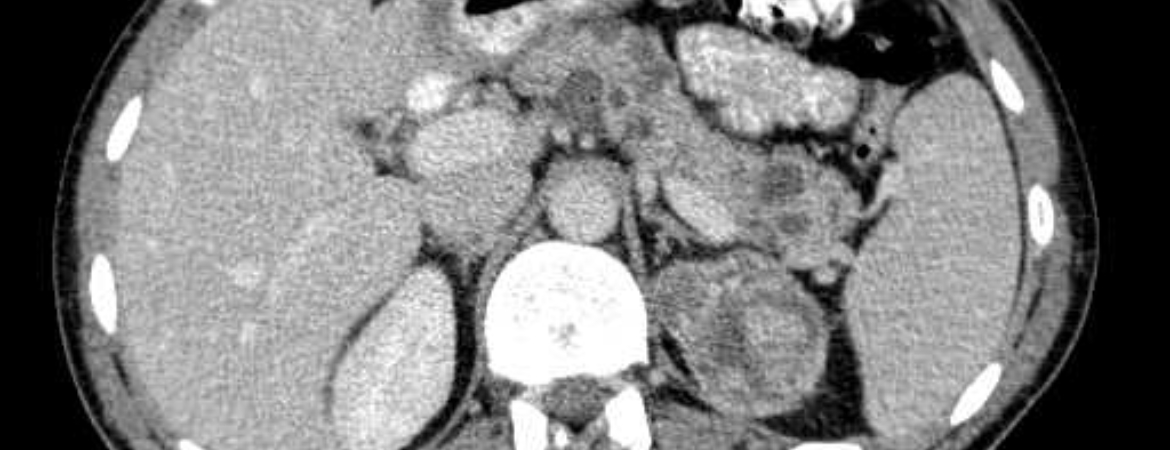
Les carcinomes rénaux sont souvent la seule entité tumorale maligne dans la maladie de VHL. Ils sont fréquemment multiples et bilatéraux et sont exclusivement de type à cellules claires sur le plan histologique. Une opération devrait être envisagée pour les tumeurs de >3 cm. Si une résection est nécessaire en raison de la croissance ou de la localisation de la tumeur, il convient d’opter pour une procédure chirurgicale préservant le néphron. Dans certains centres, l’opération n’est recommandée qu’à partir de 4 cm. L’ablation par micro-ondes, l’ablation par radiofréquence ou la cryothérapie ne sont envisagées que dans des cas particuliers, en fonction de la localisation et de la taille de la tumeur. La perte de la fonction rénale ne peut pas toujours être évitée et des thérapies de remplacement rénal ou une transplantation rénale peuvent être indiquées avec le temps. En cas de carcinome rénal associé à la maladie de VHL, une transplantation est certes possible en principe, mais elle doit être évaluée de manière critique en raison du risque de métastases et du risque de tumeurs secondaires (en particulier phéochromocytomes et tumeurs neuroendocrines). Une discussion compétente au sein de l’équipe multidisciplinaire est nécessaire à cet égard.
Les phéochromocytomes sont souvent asymptomatiques, en particulier lorsqu’ils sont de petite taille. Les symptômes sont dus à la concentration accrue de catécholamines libérées par la tumeur et la maladie peut entre autres s’accompagner d’hypertension, de tachycardie, de palpitations et de troubles du rythme cardiaque. En outre, des symptômes non spécifiques, tels que céphalées, flush, agitation, accès de transpiration, hyperglycémie, perte de poids ou fatigue, peuvent survenir. Le traitement de choix est la résection chirurgicale de la tumeur. Etant donné que les phéochromocytomes peuvent apparaître de manière synchrone ou métachrone dans les deux glandes surrénales dans le cadre de la maladie de VHL et qu’ils sont le plus souvent de nature bénigne, une résection épargnant les glandes surrénales peut être envisagée, en fonction de la localisation et de la taille du phéochromocytome, afin d’éviter une insuffisance surrénale à moyen terme. Outre les phéochromocytomes, la maladie de VHL peut également se manifester par des paragangliomes.
Des lésions pancréatiques surviennent au fil du temps chez une grande partie des personnes atteintes de la maladie de VHL. Les principales lésions sont des kystes pancréatiques ou des cystadénomes séreux. Ces derniers sont généralement bénins et ne présentent pas de tendance à la dégénérescence. Des tumeurs neuroendocrines (TNE) pancréatiques, qui ne sont le plus souvent pas actives sur le plan hormonal, peuvent survenir en tant que lésions solides. En cas d’extension de ≥3 cm ou de croissance rapide, une indication opératoire doit être envisagée de manière interdisciplinaire. En cas de métastases ou d’impossibilité de résection, il est parfois nécessaire de recourir à un traitement systémique.
Confirmation du diagnostic
En cas de suspicion de maladie de VHL et de critères cliniques remplis, il convient de procéder à un test génétique moléculaire pour détecter des variants du gène VHL, après un conseil génétique. Un variant pathogène du gène VHL peut être détecté chez une grande partie des patientes et patients chez lesquels les critères cliniques sont remplis. En cas de mise en évidence d’une mutation de la lignée germinale, les descendants devraient également être orientés vers un conseil génétique. Dans la mesure où certaines lésions de la maladie de VHL se manifestent déjà pendant l’enfance mais peuvent rester longtemps asymptomatiques, le dépistage devrait commencer dès l’enfance.
Prise en charge interdisciplinaire en cas d’une mutation du gène VHL
La prise en charge des porteuses et porteurs d’une mutation du gène VHL et des personnes atteintes de la maladie de VHL représente un défi et devrait être assurée de manière interdisciplinaire par une équipe expérimentée. Les personnes concernées courent le risque de développer une ou plusieurs lésions associées à la maladie de VHL tout au long de leur vie; elles savent qu’elles devront peut-être subir des interventions multiples et lourdes et s’inquiètent des possibles limitations physiques.
A l’hôpital cantonal de Saint-Gall, une grande cohorte de personnes atteintes de la maladie de VHL est déjà prise en charge depuis de nombreuses années par une équipe multidisciplinaire qui coordonne les examens de dépistage réguliers nécessaires (tab. 3 et 4), assure une transition de la pédiatrie vers la médecine pour adultes (consultation pour adolescents) et propose un conseil génétique. De plus, l’étroite collaboration interdisciplinaire et les conférences régulières (RCP dédié à la maladie de VHL et RCP spécifiques aux organes) permettent un suivi individuel de chaque personne concernée. L’objectif est d’assurer un suivi efficace et étroit, de sorte que des mesures et des traitements puissent être mis en place précocement en cas de nouvelles lésions.
Tableau 3: Aperçu des examens annuels de dépistage chez les adultes atteints de la maladie de VHL (à partir de 18 ans)
Examen
Anamnèse, examen clinique avec signes vitaux
Analyses de laboratoire (hémogramme; chimie clinique, y compris métanéphrines)
IRM du neurocrâne, y compris rocher et neuroaxe, avec produit de contraste
IRM de l’ensemble de l’abdomen avec produit de contraste
Examen du fond d’œil
Audiométrie de base, puis annuellement en cas de TSEL
IRM: imagerie par résonance magnétique; TSEL: tumeurs du sac endolymphatique; VHL: von Hippel-Lindau.
Le tableau 3 doit être considéré comme des recommandations. En fonction des symptômes et des résultats, le dépistage doit être adapté et la fréquence des examens doit être augmentée si nécessaire.
Tableau 4: Aperçu des examens annuels de dépistage chez les enfants atteints de la maladie de VHL (en fonction de l’âge)
Examen
Age
Anamnèse, examen clinique avec signes vitaux
Début le plus tôt possible
Analyses de laboratoire (hémogramme; chimie clinique, y compris métanéphrines)
A partir de 8–10 ans (dès que la prise de sang est possible sans stress pour l’enfant, alternativement détermination des métanéphrines dans l’urine)
IRM du neurocrâne, y compris rocher et neuroaxe, avec produit de contraste
A partir de 12 ans
IRM de l’ensemble de l’abdomen avec produit de contraste
A partir de 12–16 ans, éventuellement plus tôt en fonction du risque.A partir de 8–10 ans, échographie de l’abdomen par un radiologue pédiatrique expérimenté.
Examen du fond d’œil
A partir de 5–6 ans
Audiométrie
Lors de la pose du diagnostic et en cas de symptômes ou de TSEL à l’IRM, à tout moment comme référence. Uniquement en cas de TSEL: annuellement
IRM: imagerie par résonance magnétique; TSEL: tumeurs du sac endolymphatique VHL: von Hippel-Lindau.
Le tableau 4 doit être considéré comme des recommandations. En fonction des symptômes et des résultats, le dépistage doit être adapté et la fréquence des examens doit être augmentée si nécessaire.
Outre le suivi médical étroit, les personnes concernées ont également la possibilité de faire enregistrer leurs antécédents médicaux dans un registre de la maladie de VHL. Ce registre permet d’obtenir une vue d’ensemble de l’étendue de la maladie de VHL dans l’espace germanophone. Il peut également offrir des possibilités de participation à des études.
Le belzutifan en tant que nouvelle option thérapeutique
Les options thérapeutiques systémiques pour les lésions associées à la maladie de VHL qui ne sont pas éligibles à un traitement local ou pour lesquelles le traitement local est associé à des complications potentiellement graves sont limitées.
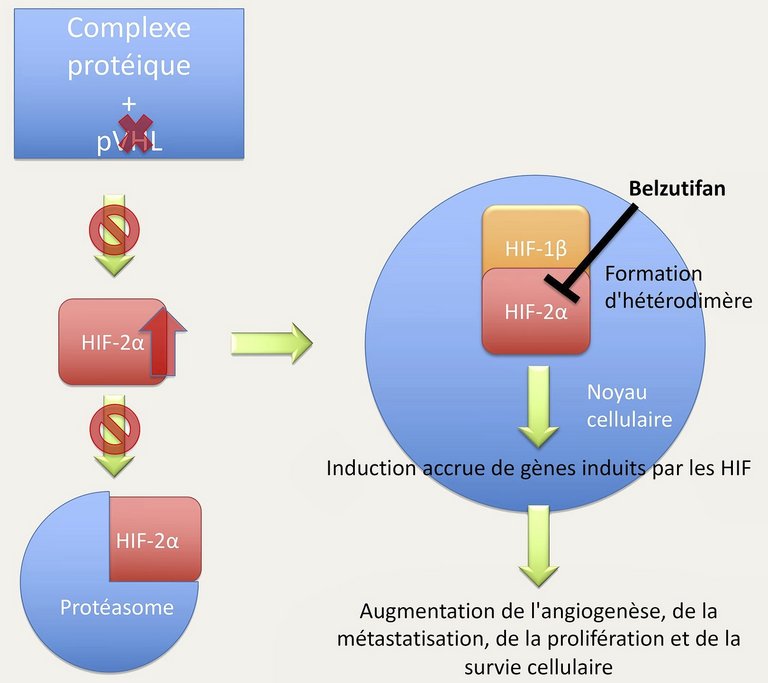
La pathogenèse de la maladie de VHL a fait l’objet de recherches intensives au cours des dernières décennies [5]. Les variants pathogènes de la lignée germinale dans le gène VHL résultent en une perte de fonction de l’une des deux copies/allèles du gène VHL. La perte du deuxième allèle est nécessaire pour qu’il y ait une perte complète de la fonction de la pVHL et donc un développement tumoral. La pVHL forme un complexe avec d’autres protéines, qui contrôle la dégradation de ses protéines cibles. Les protéines HIF sont des facteurs de transcription dont la dégradation est influencée par la pVHL. Les gènes régulés par les HIF ont à leur tour une influence essentielle sur des processus clés du métabolisme cellulaire [6]. Chez les personnes atteintes de la maladie de VHL, l’absence de dégradation des HIF sous le contrôle de la pVHL entraîne une activation excessive des gènes régulés par les HIF, qui favorisent le développement de tumeurs. Ces dernières années, il apparaît de plus en plus clairement que le HIF-2α joue un rôle déterminant dans le développement des carcinomes rénaux, mais aussi dans celui d’autres lésions associées à la maladie de VHL [7].
Avec le développement du belzutifan (MK-6482), un inhibiteur de HIF-2α, il existe désormais pour la première fois une option thérapeutique ciblée (fig. 1). Dans le cadre d’une étude de phase II ouverte à un seul bras portant sur 61 patientes et patients atteints de la maladie de VHL et d’un carcinome rénal, une réponse objective (critère d’évaluation primaire de l’étude) de 49% a pu être documentée selon les critères RECIST («Response Evaluation Criteria In Solid Tumors»), et un rétrécissement tumoral a globalement pu être constaté chez 92% des patientes et patients participant à l’étude [8]. D’autres lésions associées à la maladie de VHL ont également répondu (tumeurs du pancréas 77%; TNE pancréatiques 91%; hémangioblastomes 30%), et ce avec une bonne tolérance au traitement. De même, une stabilisation ou une amélioration a été démontrée chez 100% des patientes et patients avec atteinte rétinienne. Les effets indésirables fréquents du belzutifan sont l’anémie, la fatigue, les vertiges, les céphalées, les nausées et la dyspnée. En résumé, le développement du belzutifan offre pour la première fois une option thérapeutique ciblée pour les patientes et patients atteints de carcinome rénal et d’autres altérations associées à la maladie de VHL.
Figure 1: Les HIF-1β et HIF-2α forment un hétérodimère dans le noyau cellulaire et entraînent ainsi l’induction de leurs gènes cibles. En cas de pVHL défectueuse, les HIF ne sont plus dégradés de manière simplifiée par le protéasome en raison de l’absence d’ubiquitination par la pVHL. En tant qu’inhibiteur de HIF-2α, le belzutifan empêche la formation nécessaire de l’hétérodimère et donc l’induction des gènes induits par les HIF.
HIF: hypoxia-inducible factor; pVHL: la protéine de von Hippel-Lindau.
Jusqu’à présent, le belzutifan n’est pas encore autorisé en Suisse, mais les personnes concernées devraient bientôt y avoir accès via l’art. 71c de l’Ordonnance sur l’assurance-maladie (OAMal). Aux Etats-Unis, il existe désormais une autorisation de la «Food and Drug Administration» (FDA) pour le belzutifan en cas de carcinome rénal associé à la maladie de VHL, d’hémangioblastome du SNC ou de TNE pancréatique ne nécessitant pas de traitement chirurgical immédiat. Au Royaume-Uni, le belzutifan est désormais autorisé en cas de carcinome rénal à cellules claires associé à la maladie de VHL.
Perspectives
Le développement du belzutifan offre une nouvelle option de traitement de la maladie de VHL, dont la disponibilité en Suisse est attendue prochainement. En plus des preuves existantes de l’efficacité dans le carcinome rénal, d’autres lésions associées à la maladie de VHL montrent une réponse au belzutifan, avec un profil d’effets indésirables favorable. Jusqu’à présent, il n’a pas encore été évalué si le traitement par belzutifan permettait même de prévenir l’apparition de nouvelles lésions. Le développement de mécanismes de résistance fait actuellement l’objet de recherches. On peut néanmoins s’attendre à ce que le belzutifan, en tant que monothérapie bien tolérée, modifie le traitement des personnes concernées. L’un des défis posés par le traitement par belzutifan sera certainement de déterminer le début optimal du traitement et, en cas de bonne réponse, de ne pas rater le moment où un traitement local sera possible.
En vue d’une prise en charge moderne des patientes et patients, une consultation vidéo est prévue en complément des consultations cliniques à l’hôpital cantonal de Saint-Gall, afin de minimiser l’investissement en temps pour les personnes concernées et d’augmenter l’adhérence à la prévention et au traitement. En particulier en cas de longs trajets pour se rendre à l’hôpital ou de séjours à l’étranger, la mise en place de nouvelles structures de soins par télémédecine permet d’optimiser et d’adapter individuellement le suivi étroit des patientes et patients déjà connus.
L’essentiel pour la pratique
La maladie de von Hippel-Lindau (VHL) est une maladie multisystémique qui s’accompagne de la survenue de différentes tumeurs bénignes et malignes.
La survenue d’un carcinome rénal associé à la maladie de VHL est déterminante pour le pronostic et est particulièrement éprouvante pour les personnes concernées.
La prise en charge des personnes concernées devrait être assurée par une équipe interdisciplinaire et nécessite des examens de dépistage réguliers, centralisés et coordonnés.
Le belzutifan est le premier traitement systémique ciblé à être autorisé aux Etats-Unis et au Royaume-Uni. Il est efficace non seulement pour les carcinomes rénaux, mais aussi pour d’autres lésions associées à la maladie de VHL.
Kira-Lee Koster
Klinik für Medizinische Onkologie und Hämatologie, Kantonsspital St. Gallen, St. Gallen
Disclosure statement
CR: honoraires de consultance à l'institut de Pfizer, Bristol-Myers Squibb, MSD Oncology, Bayer Schweiz et IPSEN. IB: subventions à l'institut pour des contributions d’experts au MSD Forum. DE: honoraires de consultant de MSD Sharp Dohme. IK: honoraires de conférence de Amgen/UCB. TH: subvention de Bayer Schweiz et de l’association allemande VHL; consultant pour Alexion, Sanofi, Amicus et MSD. Les autres auteurs ont déclaré ne pas avoir de conflits d’intérêts potentiels.
Correspondance
Kira-Lee Koster
Klinik für Medizinische Onkologie und Hämatologie
Kantonsspital St. Gallen
Rorschacher Strasse 95, CH-9007 St. Gallen
Références
1 Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet. 2003;361(9374):2059–67.
2 Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, Ferguson-Smith MA. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77(283):1151–63.
3 Schmid S, Gillessen S, Binet I, Brändle M, Engeler D, Greiner J, et al. Management of von hippel-lindau disease: an interdisciplinary review. Oncol Res Treat. 2014;37(12):761–71.
4 Nielsen SM, Rhodes L, Blanco I, Chung WK, Eng C, Maher ER, et al. Von Hippel-Lindau disease: genetics and role of genetic counseling in a multiple neoplasia syndrome. J Clin Oncol. 2016;34(18):2172–81.
5 Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22(24):4991–5004.
7 Barry RE, Krek W. The von Hippel-Lindau tumour suppressor: a multi-faceted inhibitor of tumourigenesis. Trends Mol Med. 2004;10(9):466–72.
8 Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for renal cell carcinoma in von Hippel-Lindau disease. N Engl J Med. 2021;385(22):2036–46.
9 vhl.org [Internet]. Boston, MA, USA: The VHL Alliance; c2022 [cited 2022 Oct 19]. Verfügbar unter: https://www.vhl.org/