La mucoviscidose héréditaire homozygote récessive est la maladie génétique avec réduction de l’espérance de vie la plus fréquente dans la population caucasienne. Au cours des 20 dernières années, le perfectionnement des stratégies diagnostiques et thérapeutiques a permis d’améliorer considérablement aussi bien la qualité de vie des personnes concernées que le taux moyen de survie. Cette tendance persiste grâce à des innovations telles que l’introduction du dépistage néonatal et des traitements spécifiques aux mutations, mais présente également de nouveaux défis en termes de prise en charge clinique.
Spectre des caractéristiques génétiques et cliniques
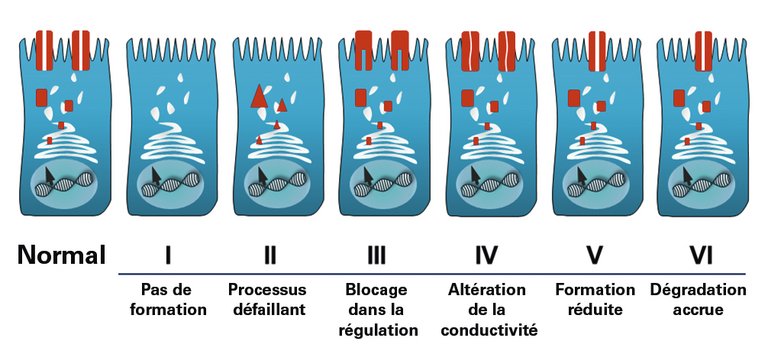
La mucoviscidose (MV) est causée par une mutation au niveau d’un gène situé sur le bras long du chromosome n° 7, qui encode la protéine «cystic fibrosis transmembrane conductance regulator»(CFTR). La protéine CFTR forme le canal ionique de chlorure à la surface des cellules épithéliales humaines. Par conséquent, chez les individus présentant une mutation homozygote ou hétérozygote composite au niveau du gène CFTR, la formation ou la fonction du canal chlorure sont fondamentalement perturbées, bien que seulement env. 10% des >2000 mutations du CFTR connues jusqu’à présent déclenchent avec certitude la maladie. En fonction de la nature du trouble de la synthèse de la protéine CFTR, les mutations du gène CFTR sont classées dans six catégories présentant des conséquences différentes sur la fonctionnalité du canal chlorure (fig. 1). Les porteurs hétérozygotes du gène sont sains (prévalence en Europe centrale env. 1:25). Outre le phénotype de la MV «classique» avec une manifestation multiorganique, des symptômes et complications typiques (tab. 1), des mutations du CFTR s’accompagnant d’une forme plus modérée de la maladie, oligosymptomatique ou monosymptomatique, sont connues. Les symptômes peuvent alors en partie survenir pendant l’enfance ou seulement après, et se manifestent souvent sous forme de pancréatite, sinusite, polypes nasaux, bronchectasie diffuse et/ou infertilité chez l’homme. Les personnes concernées ne présentent généralement aucune insuffisance pancréatique ni aucune pneumopathie grave. C’est pourquoi il est essentiel d’envisager un dysfonctionnement du CFTR également chez les adultes présentant des symptômes suggestifs pour lesquels aucune autre cause n’a pu être trouvée. Ces phénotypes autrefois qualifiés de MV «atypique», dont la manifestation et le caractère diffèrent, sont aujourd’hui appelés «CFTR-related disorders» (CFTR-RDs) suite à un consensus de la «European Cystic Fibrosis Society» (ECFS) [1]. Un aperçu actualisé en permanence des mutations connues du CFTR déclenchant une MV ainsi que de leurs fonctionnalités et manifestations cliniques se trouvent sur le site Web du projet CFTR2 (https://cftr2.org).
Figure 1: Classes de mutation du CFTR avec dysfonctionnement résultant du canal chlorure (avec l’aimable autorisation de Helge Hebestreit, Würzburg. Représentation originale dans: Speer-Gahr: Pädiatrie. VII. Zystische Fibrose. 2013. pp 499–505 [p501]. H. Hebestreit, A. Hebestreit. Springer-Verlag Berlin Heidelberg. Avec l’aimable autorisation de Springer Nature).
Tableau 1:Symptômes cliniques et complications de la mucoviscidose (MV) en fonction de l’âge. Les pourcentages entre parenthèses donnent la prévalence en Suisse en 2014 pour autant que les données soient disponibles (d’après le rapport annuel 2014 ECFSPR).
Age
Présentation clinique
Respiratoire
Gastro-intestinale et autres
Indépendamment de l‘âge
– Toux productive – Production d’expectorations – Infections des voies respiratoires avec pathogènes typiques (Staphyloccocus aureus, Haemophilus influenzae, Pseudomonas aeruginosa)
– Stagnation du poids – Stéatorrhée – Diarrhée – Ballonnements – Douleurs abdominales – Carence en vitamines liposolubles – Alcalose métabolique hypochlorémique (syndrome pseudo-Bartter, syndrome de perte de sel)
Comme les enfants, plus: – Bronchiectasies – Hémoptysies – Pneumothorax – Aspergillose bronchopulmonaire allergique (ABPA, 5,7%) – Infection par mycobactéries atypiques – Hippocratisme digital, ongles en verre de montre – Rhinosinusite chronique – Polypose naso-sinusienne – Anosmie
Comme les enfants, plus: – Pancréatite aiguë – Hépatopathie (23,0%) – Hypertension portale – Diabète sucré associé à MV (DAMV; 23,7% pour les ≥18 ans) – Ostéoporose – Arthrite associée à la MV – Absence congénitale bilatérale des canaux déférents (CBAVD)
ECFSPR = «European Cystic Fibrosis Society Patient Registry»
Etablissement du diagnostic hier et aujourd’hui
«Malheur à l’enfant chez qui un baiser sur le front a un goût salé. Il est ensorcelé et doit mourir bientôt.» Ce proverbe du MoyenÂge transmis par les sages-femmes en Europe du Nord établissait déjà autrefois un rapport diagnostique entre une teneur élevée en sel de la transpiration et une maladie létale encore inconnue. De nos jours, l’établissement du diagnostic de la MV repose sur des symptômes cliniques typiques et la confirmation biochimique du dysfonctionnement sous-jacent de la protéine CFTR. La détermination de la concentration de chlorure dans la sueur par ionophorèse quantitative à la pilocarpine («test de la sueur») constitue le standard universel du diagnostic du dysfonctionnement du CFTR [2]. La méthode renvoie à la description de Gibson et Cooke en 1959, sachant que la collecte de la sueur a été simplifiée sur le plan technique au fil des décennies, le système Macroduct® disponible dans le commerce étant principalement utilisé aujourd’hui en Suisse. Ces dernières années, outre la mesure de la conductivité de la sueur, une autre méthode s’est développée et établie, bien qu’elle soit uniquement acceptée en tant que méthode de dépistage selon les recommandations internationales actuellement en vigueur, tandis qu’en cas de résultat anormal au test, la détermination confirmatoire de la concentration de chlorure doit être effectuée. Ici aussi, des systèmes fiables de test sont disponibles dans le commerce, le système Nanoduct® étant largement répandu en Suisse. Le test de la sueur en deux étapes (dépistage au moyen de la conductivité, confirmation par le chlorure uniquement en cas de conductivité accrue) est entre-temps réalisé dans de nombreux laboratoires, puisqu’en cas de conductivité normale, une MV peut être en grande partie exclue. D’autres méthodes telles que la détermination de l’osmolalité de la sueur ou la seule mesure de la concentration de sodium ou de potassium sans chlorure sont aujourd’hui obsolètes. Lors de l’interprétation des résultats du test de la sueur, il convient d’observer que les valeurs de référence de la mesure de conductivité sont supérieures à celles de la concentration de chlorure, puisque la conductivité est également déterminée par d’autres ions que le chlorure (tab. 2). Le laboratoire pratiquant le test de la sueur doit documenter, outre la méthode employée, la plage de valeurs de référence, le volume et le poids de la sueur recueillie ainsi que, en cas de mesure de la conductivité, le taux de transpiration avec les valeurs limites correspondantes. Le résultat d’un test avec une quantité ou un taux trop faible de sueur ne doit pas être utilisé et nécessite de répéter l’analyse.
Tableau 2:Valeurs de référence pour les différentes méthodes de test de la sueur.
Concentration de chlorure
Conductivité (équivalents NaCl)
MV
≥60 mmol/l
≥80 mmol/l
Zone grise
30–59 mmol/l
50–79 mmol/l
Pas de MV
<30 mmol/l
<50 mmol/l
Volume de sueur
≥15 µl (Macroduct®)
≥3 µl (Nanoduct®)
Taux de sueur
./.
≥1,0 g/m2/min
MV = mucoviscidose, ./. = non applicable
Outre une constellation symptomatique suggestive, la présence d’une MV chez un frère ou une sœur ou encore un dépistage néonatal positif peuvent également représenter une indication en vue de réaliser un test de la sueur chez les individus asymptomatiques. Ce test doit, dans la mesure du possible, être effectué dans un centre spécialisé dans la MV. Si le test de la sueur est positif, le patient doit être immédiatement adressé au centre MV le plus proche pour un diagnostic complémentaire (fig. 2). L’établissement du diagnostic nécessite en règle générale d’être confirmé par un deuxième test de la sueur. En cas de confirmation de la MV ou d’incertitude (par ex. test de la sueur intermédiaire ou sans résultat exploitable), s’en suivent, outre l’examen de la fonction pancréatique (baisse de la lactase pancréatique dans les selles), la détermination des mutations du CFTR sur les deux allèles, ce qui n’est pas uniquement pertinent pour le pronostic de l’évolution de la maladie et pour un conseil génétique (par ex. suite du planning familial), mais comporte aussi de plus en plus souvent des conséquences thérapeutiques à l’ère des traitements spécifiques aux mutations.
Figure 2: Diagramme de flux pour l’établissement du diagnostic d’une mucoviscidose (MV) en présence de suspicion clinique (modifié et simplifié selon [2]).
CFTR = «cystic fibrosis transmembrane conductance regulator»
Dans de rares cas, les méthodes mentionnées ne permettent pas de déterminer de manière univoque si une MV est présente. La détermination de la différence de potentiel nasal (DPN) ou la mesure du courant de court-circuit au niveau de l’intestin (ICM) peuvent apporter des clarifications sur la fonction du CFTR. Ces méthodes ne sont toutefois disponibles que dans de rares centres hautement spécialisés.
Dépistage néonatal depuis 2011
Le dépistage néonatal de la MV a été introduit en Suisse le 1er janvier 2011 [3]. Il s’agit d’une procédure de test en deux étapes basé sur un prélèvement sanguin au niveau du talon réalisé au quatrième jour de vie (trypsinogène immunoréactif [TIR], si positif suivi d’une analyse de la mutation sur la base des 18 mutations du CFTR actuellement les plus fréquentes Suisse). Tous les enfants positifs sont directement adressés par le laboratoire central de dépistage, établi à l’hôpital universitaire pédiatrique de Zurich, au centre MV le plus proche pour une mise au point diagnostique complémentaire. Il y est d’abord pratiqué un test de la sueur. En cas de résultat anormal, une autre analyse de la mutation destinée à détecter les deux allèles du CFTR est pratiquée et un échantillon de selles est prélevé à la recherche de lactase pancréatique. Les résultats actuels du dépistage néonatal fournissent une incidence de la MV classique en Suisse de 1:3631 nouveau-nés (dépistage néonatal Suisse, rapport annuel 2016, téléchargement sur http://www.neoscreening.ch/fr/rapportannuel.htm). En Suisse, la sensibilité du dépistage pour la détection d’une MV classique est de 95%; cela signifie que le test présente jusqu’à 5% de résultats faussement négatifs. Ainsi, un résultat normal au dépistage n’exclut pas avec certitude une MV. C’est la raison pour laquelle, en présence de symptômes cliniques typiques (tab. 1), il convient d’envisager une MV même chez les patients présentant un résultat normal au test de dépistage néonatal et de prescrire d’autres investigations diagnostiques. Tous les nouveau-nés atteints d’iléus méconial doivent être soumis au test de la sueur ou à une analyse génétique, puisqu’il est possible que ces enfants présentent un TIR normal à la naissance et ne soient ainsi pas détectés par le biais du dépistage néonatal.
Outre les enfants atteints de MV classique, certains enfants présentant également de très légères mutations et un test de la sueur limite, c’est-à-dire une fonction résiduelle de CFTR, sont aussi obligatoirement enregistrés à la suite du dépistage néonatal. Ces enfants ne souffrent pas de MV, mais sont classés dans la catégorie «CF screen positive, inconclusive diagnosis» (CFSPID) [4]. Certains d’entre eux présentent à l’âge adulte des symptômes isolés ou combinés d’un CFTR-RD, de sorte qu’en présence d’un CFSPID, les symptômes typiques d’un dysfonctionnement du CFTR doivent par la suite être contrôlés et surveillés par les pédiatres et les médecins de famille.
Résultats tirés du registre des patients pour la prise en charge clinique
Pour l’année 2015, tous les centres MV de Suisse participent pour la première fois au «European Cystic Fibrosis Society Patient Registry» (ECFSPR). Il s’agit là du point de départ de futurs projets de référencement ayant pour objectif d’optimiser au niveau national la qualité de la prise en charge de personnes atteintes de MV en Suisse. En 2014, 789 patients atteints de MV et en provenance de la Suisse ont été enregistrés au ECFSPR. En incluant dans le calcul les nombres estimés de patients provenant des centres non participants, on peut partir du principe qu’en 2014, près de 950 personnes atteintes de MV vivaient en Suisse, environ 50% de ces personnes étant âgés de plus de 17 ans. En 2014, l’âge médian au moment du diagnostic s’élevait à seulement 6 mois (moyenne 2,7 ans), 15% des patients en vie ayant déjà été diagnostiqués dans le cadre du dépistage néonatal. Un autre déplacement vers l’âge néonatal peut être attendu du dépistage. Chez 99% des patients suisses, le génotype exact est connu. En Suisse, 86% des patients portent la mutation du CFTR la plus fréquente delF508 sur au moins un allèle, 47% sont homozygotes pour cette mutation. La fréquence est inférieure à 4% pour toutes les autres mutations. Le tableau 3 présente des données cliniques de référence issues du rapport annuel 2014 du ECFSPR. (Consultable sur https://www.ecfs.eu/projects/ecfs-patient-registry/annual-reports).
Tableau 3: Valeurs de référence cliniques de la mucoviscidose en fonction des groupes d’âge en Suisse en 2014 (d’après le rapport annuel 2014 ECFSPR). Les données microbiologiques portent sur la prévalence d’une infection pulmonaire chronique.
<18 ans
≥18 ans
VEMS vs. valeurs prédites1,2
89,2% (77,4, 100,5)
62,1% (46,9, 81,8)
Poids (z-scores)
–0,4 (–1,1, 0,3)
–0,5 (–1,2, 0,1)
P. aeruginosa
12,8%
53,1%
S. aureus
55,2%
49,2%
MNT3
0,8%
6,5%
1 «Global Lung Initiative equations»; tous les patients à partir de 6 ans sans transplantation pulmonaire 2 Médiane (25e, 75e percentiles) 3 MNT = mycobactéries non tuberculeuses ECFSPR = «European Cystic Fibrosis Society Patient Registry»
Le traitement dans un centre spécialisé est essentiel
Comme dans la plupart des pays européens, les personnes atteintes de MV sont, en Suisse aussi, actuellement traitées dans des centres hautement spécialisés qui proposent une prise en charge interdisciplinaire globale, pouvant mettre à disposition du personnel spécifiquement formé en conséquence et présentant de longues années d’expertise en rapport avec ce tableau clinique complexe. Les exigences auxquelles est soumis un centre MV moderne ainsi que les mesures préventives et thérapeutiques s’orientent sur la base des recommandations internationales telles que les «Standards of Care» de l’ECFS [5]. Les patients sont généralement reçus au centre tous les 3 mois, les examens étant effectués dans le cadre de programmes standardisés pouvant légèrement varier d’un centre à l’autre en fonction de l’expérience, des ressources et des conditions techniques ainsi que selon l’âge des patients (tab. 4). L’objectif de ces programmes de surveillance est d’une part d’assurer le dépistage précoce et la surveillance de l’évolution des manifestations organiques et des complications, mais d’autre part aussi de contrôler et modifier le traitement suivant l’évolution individuelle de la maladie. Des formations spécifiques des patients et de leurs proches doivent avoir lieu à intervalle régulier afin de faire face de manière efficace aux défis d’une prise en charge complexe de la maladie au quotidien en fonction des différentes phases de vie.
Tableau 4: Programme de surveillance standardisé en centre de MV (modifié selon [5]); ici à l’exemple de l’hôpital pour enfant de Zurich.
Examen
Détails
Fréquence
Instruction de physiothérapie
Y compris vérification de la technique d’inhalation; en centre ou dans un cabinet spécialisé
Hebdomadaire à bimensuelle
Anamnèse
Médecin spécialiste de la MV
Tous les 3 mois
Contrôle thérapeutique
Médecin spécialiste de la MV; y compris information sur la supplémentation en sel
Examen physique
Médecin spécialiste de la MV
Gestion de la maladie
Infirmière spécialiste de la MV
Formation des patients
Y compris hygiène; médecin spécialiste de la MV et infirmière spécialiste de la MV
Etat nutritionnel
Poids, taille, indice de masse corporelle, poids-pour-taille
Fonction pulmonaire
Spirométrie, pléthysmographie corporelle; à partir de 4 ans
«Multiple-Breath Washout»
Indice de clairance pulmonaire (LCI); à partir de 3 ans
Microbiologie
Expectorations, ou frottis pharyngé
Conseil social
Assurances, finances, droit
Prélèvements sanguins annuels
Hémogramme, électrolytes, oligoéléments, fer, ferritine, vitamines liposolubles A/D, protéine totale, albumine, enzymes hépatiques, statut de la coagulation, paramètre de cholestase, acide biliaire, glycémie à jeun, HbA1c, paramètres rénaux, ImmunoCAP (s×1, m×1)
Annuelle
Hyperglycémie provoquée per os
A partir de 10 ans
Echographie abdominale
Conseil nutritionnel
Y compris contrôle de la supplémentation enzymatique
Bronchoscopie avec LBA
Chez les patients ne produisant pas d’expectorations ou si nécessaire
Dépistage MNT
A partir du LBA ou des expectorations; à partir de 10 ans
Outre les thèmes médicaux spécifiques à la maladie et au traitement, cela inclut aussi le conseil sur les questions de droit social, financières et psychologiques, de même qu’en termes d’hygiène, de mesure comportementale au jardin d’enfants, à l’école et lors des voyages ainsi qu’un conseil professionnel. Les jeunes parents qui souhaitent d’autres naissances et les patients adultes désirant avoir des enfants doivent bénéficier d’un diagnostic génétique approfondi du partenaire, en règle générale avec l’appui d’un centre de génétique humaine. Pour certains patients ou certaines familles, une réhabilitation interdisciplinaire stationnaire peut s’avérer pertinente afin d’optimiser la prise en charge de la maladie ou stabiliser son évolution.
Le patient adulte au cœur des préoccupations
Au vu des avancées diagnostiques et thérapeutiques modernes, presque tous les patients atteignent actuellement l’âge adulte dans les pays industrialisés. En Suisse, la moitié des personnes vivantes atteintes de MV sont âgées de plus de 17 ans. En 2012, l’âge de survie moyen dans divers pays industrialisés se trouvait entre 40 et 50 ans [6]. Par conséquent, la transition des patients devenus adultes du centre MV pédiatrique vers un centre pour adultes est explicitement requise par la ECFS [5] – elle est actuellement établie dans toute la Suisse. Bien que la ECFS ait émis des recommandations claires sur la structure et le déroulement, les programmes de transition varient en réalité fortement d’un centre à l’autre en termes de structure et de contenu. Des données issues du registre américain de la MV suggèrent une supériorité de la transition vers un centre pour adultes par rapport à l’usage encore souvent en pratique de poursuivre l’accompagnement des patients adultes au centre pédiatrique, du moins en ce qui concerne la fonction pulmonaire [7]. Par ailleurs, la transition est également pertinente du point de vue clinique: avec l’âge, les adultes atteints de MV souffrent d’affections internes telles qu’hypertension artérielle ou tumeurs, qui sont souvent indépendantes de la MV mais susceptibles de la compliquer. Un savoir-faire en médecine interne est alors requis, ce qui dépasse généralement les compétences de la pédiatrie. Du fait de ces modifications démographiques au sein de la population atteinte de MV, les structures de prise en charge médicale doivent donc également évoluer: des centres MV interdisciplinaires de médecine interne doivent être créés; les disciplines qui n’étaient jusqu’à présent pas familières avec la MV, comme par exemple la gynécologie et l’obstétrique, doivent faire face aux exigences complexes de patients atteints de MV; le transfert des coûts depuis l’assurance invalidité jusqu’aux caisses d’assurance-maladie doit être anticipé; des thèmes tels que le plan de carrière professionnelle, le maintien à long terme de la capacité de travailler et le planning familial sont mis en avant et doivent être adressés. La planification du passage vers l’âge adulte avec la MV débute aujourd’hui dès l’enfance: tandis que l’objectif était jusqu’à récemment d’atteindre si possible l’âge adulte, les familles ayant des enfants souffrant de MV se préoccupent aujourd’hui de thèmes tels que l’éducation scolaire et la formation professionnelle ou encore le financement d’un appartement pour l’enfant devenu adulte.
Traitements innovants et établis
Ce développement démographique a été rendu possible d’une part par les programmes de surveillance établis, d’autre part en particulier aussi par le succès du traitement moderne spécifique à la MV. Celui-ci repose essentiellement sur trois piliers: le traitement enzymatique de substitution et la nutrithérapie, la sécrétolyse quotidienne par inhalation et par kinésithérapie respiratoire ainsi qu’une stratégie antibiotique agressive. Le tableau 5 présente un aperçu des principaux traitements et substances. La suite de l’article aborde brièvement les nouveaux développements des dernières années.
Tableau 5: Stratégies thérapeutiques pour le traitement de la mucoviscidose (MV) incluant les données disponibles relatives à la fréquence en Suisse en 2014 (d’après le rapport annuel 2014 ECFSPR).
Stratégie
Formes thérapeutiques
Principaux objectifs thérapeutiques
Fréquence en suisse1
Traitement nutritionnel
Enzymes pancréatiques
Développement normal du poids et de la taille
88,9%
Alimentation hautement calorique
./.2
Vitamines liposolubles
Prévention de l’hypovitaminose, carence en vitamine K et ostéoporose
./.
Supplémentation orale en chlorure de sodium
Prévention d’un syndrome de perte de sel
./.
Traitement des complications gastro-intestinales
Acide ursodésoxycholique
Traitement de la cholestase; prévention d’une hépathopathie
29,8%
Insuline
Traitement du DAMV
23,7%3
Macrogol
Traitement et prévention du SOID
./.
Sécrétolyse
Physiothérapie respiratoire (avant tout drainage autogène et aide à la sécrétolyse)
Amélioration de la clairance mucociliaire; prévention des atélectasies, bronchiectasies et infections pulmonaires / exacerbations
./.
Inhalation d’une solution hypertonique chlorure de sodium
60,9%
Inhalation de rhDNAse
38,2%
Bronchodilatation
Bêta-2-mimétiques (à courte et longue durée d’action)
Traitement préliminaire avant solution hypertonique chlorure de sodium; traitement du trouble respiratoire obstructif chronique
88,7%
Traitement de l’infection pulmonaire
Antibiotiques systémiques
Traitement de l’exacerbation pulmonaire; prévention d’une perte de la fonction pulmonaire
Antibiotiques inhalés
Eradication de P. aeruginosa; suppression en cas d’infection chronique à pseudomonas pour la prévention des exacerbations
34,7%
Macrolides (avant tout azithromycine en cas d’infection chronique à pseudomonas)
Amélioration de la fonction pulmonaire (effet anti-inflammatoire supposé)
28,3%
Antimycosiques systémiques
Traitement de l’ABPA
./.
Corticoïdes systémiques
./.
Clairance des sécrétions naso-sinusiennes
Rinçage nasal, inhalation sinusienne
Prévention / traitement de la polypose naso-sinusienne
./.
Correction du défaut de base
Correcteurs et potentiateurs de CFTR
Amélioration / stabilisation de la pneumopathie; prise de poids
<1%
1 en % de tous les patients MV 2 non documenté 3 chez les patients ≥18 ans MV = mucoviscidose, ECFSPR = «European Cystic Fibrosis Society Patient Registry», DAMV = diabète associé à la fibrose kystique, SOID = obstruction intestinale distale, ABPA = aspergillose broncho-pulmonaire allergique
Autrefois courante, la thérapie sécrétolytique par inhalation avec une solution saline physiologique a été entièrement remplacée par des solutions salines concentrées entre 3 et 7%, dont les processus osmotiques permettent le dégagement considérablement plus efficace des sécrétions. Le mannitol (Bronchitol®), un mucolytique alternatif à inhaler, a trouvé sa place dans l’arsenal thérapeutique, bien qu’il ne soit pas encore autorisé en Suisse. Avec l’apparition de la solution saline hypertonique, l’utilisation de la rhDNAse (Pulmozyme®), qui présente également une action sécrétolytique due à un processus enzymatique mais est beaucoup plus onéreuse, a quelque peu régressé, bien que des études montrent que l’effet thérapeutique de la rhDNAse sur la fonction pulmonaire s’avère supérieur à celui de la solution saline hypertonique [8].
En raison du vieillissement des patients, la colonisation pulmonaire chronique par un ou plusieurs germes problématiques tels que les souches multirésistantes de Pseudomonas, Burkholderia cepacia ou Mycobacterium abscessus joue un rôle de plus en plus crucial. Tandis qu’en Suisse, la prévalence de la colonisation pulmonaire chronique à Pseudomonas n’est encore qu’à 12,8% chez les enfants et les adolescents, elle s’élève à 53,1% chez les adultes (tab. 3). Chez les patients jeunes, une infection par un germe problématique peut souvent encore être éradiquée par des antibiotiques conventionnels administrés par voie intraveineuse ou par inhalation. En particulier pour les patients présentant une infection chronique à Pseudomonas, le développement de nouveaux antibiotiques est essentiel pour parvenir à supprimer efficacement l’activité bactérienne et prévenir par la suite des exacerbations pulmonaires et une perte de la fonction pulmonaire. Des stratégies par inhalation sont en premier lieu employées à cet effet, avec une utilisation intermittente ON/OFF par périodes de 4 semaines en tant que traitement de longue durée, parfois combiné. Outre les substances dont l’efficacité contre les Pseudomonas est établie, telles que la colistine et la tobramycine, l’aztréonam (Cayston®) a été autorisé il y a quelques années en Suisse. En ce qui concerne la tobramycine, une poudre sèche est depuis peu disponible en plus de l’inhalation humide (TOBI® Podhaler®).
Depuis peu, de nouvelles stratégies thérapeutiques s’apprêtent à potentiellement révolutionner l’évolution et le pronostic de la MV. Contrairement à la thérapie génique, qui avait éveillé un fort espoir dans les années 1990 et n’est jusqu’à présent pas parvenu à s’imposer, ces traitements visent une modulation du canal CFTR au niveau protéique, avec une distinction entre les correcteurs (meilleure disponibilité de la protéine CFTR fonctionnelle dans la membrane cellulaire) et les potentiateurs (amélioration de l’ouverture du canal et de la fonctionnalité). Le mécanisme d’action est spécifique pour une ou plusieurs classes de mutation du CFTR, d’où l’utilisation spécifique à la mutation chez les patients.
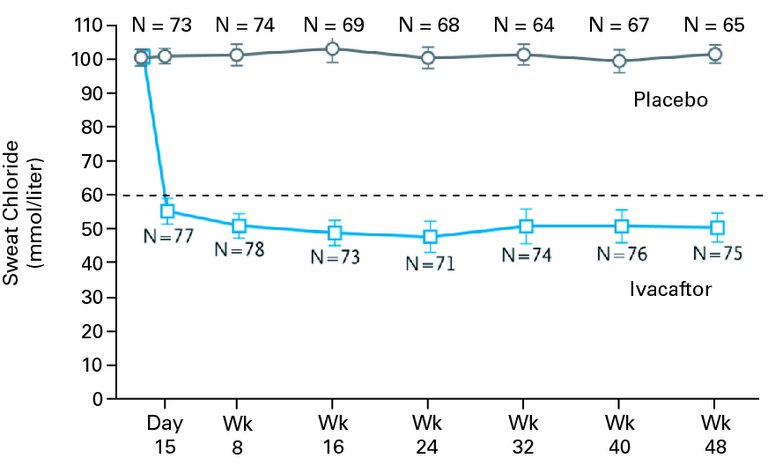
Avec l’ivacaftor (Kalydeco®), le premier potentiateur de la protéine CFTR pour certaines mutations de classe III (mutations dites «gating») est autorisé en Suisse. L’ivacaftor provoque une ouverture du canal chlorure et augmente ainsi la fonctionnalité de la protéine CFTR. Des études cliniques montrent, sous ivacaftor, une amélioration du test de la sueur d’env. 50 mmol/l, une stabilisation de l’état clinique ainsi qu’une hausse du FEV1 de près de 10% ([9], fig. 3). Cet effet sur la fonction pulmonaire dépasse très nettement les approches thérapeutiques symptomatiques conventionnelles telles que les sécrétolytiques et antibiotiques. Par ailleurs, l’ivacaftor a des répercussions positives sur l’évolution du poids, indépendamment de l’âge [9, 10]. Les expériences cliniques recueillies depuis l’autorisation de mise sur le marché confirment les données d’études, bien qu’en Suisse, seul un nombre très restreint de patients puisse profiter de l’ivacaftor du fait de la faible fréquence des mutations «gating». En raison des coûts élevés approchant les 300 000 CHF par patient et par an, la remise est assortie d’une limitation qui surveille l’efficacité pour chaque patient de manière individuelle.
Deuxième substance du groupe des modulateurs de CFTR, l’association ivacaftor/lumacaftor (Orkambi®) a été autorisée en 2015 aux Etats-Unis et en Europe ainsi qu’en 2016 en Suisse pour les patients atteints de MV présentant une mutation delF508 homozygote. Le correcteur de classe II lumacaftor améliore le pliage de la protéine CFTR, de sorte qu’une moindre quantité de protéines CFTR défectueuse soit décomposée dans les cellules épithéliales et que plus de canaux chlorure soient disponibles au niveau de la surface cellulaire. L’ivacaftor a pour fonction d’optimiser davantage la fonctionnalité des canaux. Des études cliniques montrent toutefois un effet moindre sur la concentration de chlorure dans la sueur et sur la fonction pulmonaire (env. 3% d’amélioration du FEV1) par rapport à l’effet de l’ivacaftor en cas de mutations «gating». Néanmoins, le taux d’exacerbations pulmonaires diminue à long terme de jusqu’à 40%, ce qui rend Orkambi® intéressant en particulier pour les patients atteints de MV présentant une évolution pulmonaire instable [11]. En outre, la fonction pulmonaire semble se stabiliser à moyen terme, à savoir que la baisse de la fonction pulmonaire attendue du fait de la maladie s’avère réduite au fil du temps. L’expérience clinique recueillie jusqu’à présent montre d’une part une réduction effective des exacerbations pulmonaires et une prise de poids sous Orkambi® chez certains patients avec pneumopathie avancée, mais d’autre part également une réponse hétérogène des patients, de sorte qu’il n’est actuellement pas tout à fait clair quels patients profitent le mieux du traitement.
D’autres modulateurs de CFTR sont actuellement à l’essai dans des études cliniques de phases I, II et III. Cela inclut également le développement d’une autre classe de substances destinées à renforcer l’action des correcteurs et potentiateurs (appelées «amplificateurs»). L’avenir pourrait résider dans une association de plusieurs substances, adaptée individuellement a chaque patient. Il est possible d’espérer que, dans les années à venir, des traitements efficaces spécifiques aux mutations soient mis à la disposition d’un grand nombre de patients atteints de MV.
Stratégies thérapeutiques en début et fin d’évolution de la maladie
Les nourrissons et les enfants en bas âge qui ont été diagnostiqués par un dépistage néonatal positif représentent un nouveau défi du point de vue thérapeutique. L’objectif du dépistage néonatal est d’enregistrer précocement les nourrissons atteints de MV et d’insuffisance pancréatique, et d’initier, dès l’âge néonatal, le traitement par enzymes pancréatiques de substitution ainsi que des vitamines liposolubles et une substitution de solution saline. Ces enfants ne présentent souvent aucun ou de très légers symptômes respiratoires durant les premières années de vie. Des données notamment issues de l’étude australienne AREST-CF montrent toutefois que des modifications structurelles typiques de la MV peuvent déjà être mises en évidence par tomodensitométrie pulmonaire durant les 2 premières années de vie, lorsque la plupart des enfants sont encore asymptomatiques [12]. En Suisse, l’étude multicentrique SCILD recherche actuellement quels facteurs précoces d’influence sont essentiels pour la suite de l’évolution de la maladie [13]. Par conséquent, même en cas de résultat normal sur le plan respiratoire, une thérapie sécrétolytique par inhalation et une kinésithérapie respiratoire sont initiées en même temps que le traitement enzymatique de substitution, et l’évolution gastro-intestinale et pulmonaire de la maladie ainsi que la colonisation microbienne des voies respiratoires sont surveillées tous les 3 mois. Un dépistage biochimique des complications et un procédé d’imagerie, tels que représentés au tableau 4, font généralement aussi déjà partie de la gestion clinique chez cette population de patients.
Malgré l’amélioration de l’espérance de vie et de la qualité de vie grâce au diagnostic précoce, la surveillance standardisée et l’amélioration des stratégies thérapeutiques, une transplantation pulmonaire représente pour de nombreuses personnes atteintes de MV et d’une pneumopathie terminale la dernière option de prolongation de la vie. Etant donné que le délai d’attente d’un nouvel organe comporte encore des mois à des années, il est pertinent de se présenter tôt au centre de transplantation pour une première évaluation et un conseil individuel. Le délai moyen d’attente d’une transplantation pulmonaire était de 404 jours en 2015 selon les données de Swisstransplant (médiane 202 jours) (Rapport annuel 2015 Swisstransplant, consultable sur https://www.swisstransplant.org/fr/swisstransplant/publications/rapports-annuels/). En 2014, 3,5% des patients vivants atteints de MV ont bénéficié d’une transplantation pulmonaire en Suisse. Le taux mondial de survie à 5 ans des enfants et adolescents ayant bénéficié d’une transplantation pulmonaire entre 1998 et 2014 se situe entre 50 et 55%, tandis qu’il s’élève à 65% chez les adultes [14]. Pour de plus amples informations sur la transplantation en cas de MV, veuillez vous référer aux ouvrages de référence publiés.
Perspectives
Malgré une amélioration considérable dans le domaine de la prévention et du traitement, l’évolution de la MV reste très hétérogène selon les cas. De graves évolutions peuvent aujourd’hui encore survenir dès l’enfance et l’adolescence, ce qui est parfois lié à un manque d’observance, mais probablement plus souvent à des facteurs inconnus modifiant la maladie. La détection de prédicteurs de l’évolution individuelle de la maladie, comme par exemple les marqueurs inflammatoires, doit être poursuivie. Actuellement, l’âge adulte est en revanche souvent caractérisé par la concurrence entre un traitement chronophage et une vie professionnelle et familiale limitée le moins possible, ce qui rend nécessaire une gestion rigoureuse du quotidien, en plus d’une stratégie médicale efficace. Pour les deux tranches d’âge, de nouveaux traitements spécifiques aux mutations peuvent apporter une contribution significative en stabilisant l’évolution de la maladie; toutefois, au vu des prix élevés de ces traitements, l’accès de toutes les personnes concernées n’est à l’avenir pas assuré. Le corps médical, les organisations de patients et la politique sont priés de trouver une voie commune qui ne refuse un traitement efficace à aucun patient.
L’essentiel pour la pratique
• La qualité de vie et l’âge de survie des personnes atteintes de mucoviscidose (MV) se sont améliorés au cours des dernières années. Une gestion optimisée ainsi que de nouveaux traitements efficaces ont permis de stabiliser les évolutions de la maladie.
• Des traitements sécrétolytiques efficaces, des antibiotiques inhalés modernes et des approches thérapeutiques spécifiques aux mutations, qui rétablissent ou améliorent la fonction du CFTR, offrent actuellement de nouvelles perspectives aux personnes concernées.
• Les résultats du registre européen de la MV, auquel la Suisse participe, permettent d’améliorer et d’harmoniser la qualité de la prise en charge au moyen du référencement.
• La part d’adultes atteints de MV s’élève entre-temps à près de 50%; il convient de tenir davantage compte des exigences et besoins spéciaux de ce groupe de patients vieillissants de plus en plus important.
• Introduit en 2011, le dépistage néonatal de la MV a permis un établissement du diagnostic et une initiation du traitement précoces, ce qui aura des répercussions positives sur l’évolution pulmonaire et gastro-intestinale de la maladie, justement en phase de croissance.
• Etant donné que la part des résultats faussement négatifs du dépistage néonatal de la MV s’élève à jusqu’à 5%, il convient, en présence de symptômes cliniques subjectifs, d’envisager une MV et d’effectuer un test de la sueur même chez les enfants présentant un résultat normal lors du dépistage neonatal.
• Il est possible que des phénotypes oligosymptomatiques ou monosymptomatiques ne se manifestent qu’à l’âge adulte; il convient alors aussi de réaliser un diagnostic de la MV en présence de soupçon d’un dysfonctionnement du CFTR.
• Les patients présentant un soupçon clinique de MV, un dysfonctionnement du CFTR ou un test de la sueur anormal doivent dans tous les cas être adressés à un centre MV pour des examens diagnostiques complémentaires et un traitement.
Disclosure statement
L’auteur a perçu des honoraires en tant que consultant de Vertex, Novartis, Gilead et Vifor.
Correspondance
Dr méd. Andreas Jung. Universitäts-Kinderspital Zürich Steinwiesstr. 75 CH-8032 Zürich andreas.jung[at]kispi.uzh.ch
Références
1 Bombieri C, Claustres M, De Boeck K, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10 Suppl 2:S86–S102.
2 De Boeck K, Wilschanski M, Castellani C, et al. Cystic fibrosis: terminology and diagnostic algorithms. Thorax. 2006;61:627–35.
3 Ruegg CS, Kuehni CE, Gallati S, et al. Neugeborenen-Screening auf Cystische Fibrose – Evaluation nach einem Jahr. Paediatrica. 2013;24:24–8.
4 Munck A, Mayell SJ, Winters V et al. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J Cyst Fibros. 2015; 14:706–13.
5 Kerem E, Conway S, Elborn S, Heijerman H. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros. 2005;4:7–26.
6 Sykes J, Stanojevic S, Goss CH, et al. A standardized approach to estimating survival statistics for population-based cystic fibrosis registry cohorts. J Clin Epidemiol. 2016;70:206–213.
7 Tuchman L, Schwartz M. Health outcomes associated with transition from pediatric to adult cystic fibrosis care. Pediatrics. 2013;132:847–53.
8 Suri R, Wallis C, Bush A, et al. A comparative study of hypertonic saline, daily and alternate-day rhDNase in children with cystic fibrosis. Health Technol Assessment. 2002;6:1–69.
9 Ramsey BW, Davies J, McElvaney N, Tullis E, Bell SC, Drevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and G551D mutation. N Engl J Med. 2011;365:1663–72.
10 Davies JC, Cunningham S, Harris WT, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4:107–15.
11 Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220–31.
12 Mott LS, Park J, Murray CP, et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax. 2012;67:509–16.
13 Mika M, Korten I, Qi W, Regamey N, Frey U, Casaulta C, SCILD study group. The nasal microbiota in infants with cystic fibrosis in the first year of life: a prospective cohort study. Lancet Respir Med. 2016;4:627–35.
14 Hayes D Jr, Glanville AR, McGiffin D, et al. Age-related survival disparity associated with lung transplantation in cystic fibrosis: An analysis of the registry of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2016;35:1108–15.