Ein 67-jähriger Patient stellte sich aufgrund von Fieber, Appetitlosigkeit, Abgeschlagenheit und Gewichtsverlust von 10 kg ambulant im Spital vor.
Fallbericht
Anamnese
Ein 67-jähriger Patient stellte sich aufgrund von Fieber, Appetitlosigkeit, Abgeschlagenheit und Gewichtsverlust von 10 kg ambulant in unserem Spital vor. Die Beschwerden dauerten seit sechs Wochen, anamnestisch waren keine lokalisierenden Symptome auszumachen.
Die persönliche Anamnese des Patienten war bis auf eine ähnliche Episode mit Fieber, Allgemeinzustandsminderung und erhöhten Entzündungsparametern vor drei Jahren unauffällig. Bereits damals wurde der Patient in unserer Klinik breit abgeklärt, auch eine Knochenmarkpunktion und ein PET-CT hatten keine wegweisenden Befunde ergeben. Im Intervall war es spontan zu einer Besserung der Symptome gekommen und der Patient fühlte sich während der letzten drei Jahre gesund und leistungsfähig. Drei Monate vor der aktuellen Zuweisung fiel während der Ferien in seinem Herkunftsland Spanien eine Ptose des linken Auges auf. Eine kranielle Computertomographie (CT) war unauffällig und auf eine probatorische Therapie mit systemischen Steroiden während zehn Tagen zeigte sich die Ptose regredient. Bei der aktuellen Präsentation nahm der Patient keine Medikamente ein und es bestanden keine Allergien. Der Patient hatte bis zur Pensionierung in der Gastronomie gearbeitet und ist Vater von zwei gesunden Kindern.
Status
Im klinischen Status fanden sich keine Auffälligkeiten, die Untersuchung von Haut und Gelenken war bland. Die vorbeschriebene Ptose war für uns nicht eindeutig objektivierbar, wurde aber vom Patienten und seinen Angehörigen noch diskret wahrgenommen. Ansonsten fanden sich auch keine neurologischen Auffälligkeiten.
Befunde
In den Laboruntersuchungen zeigte sich ein humoraler Entzündungszustand mit mässig erhöhtem CRP (40 mg/l) und einer diskret erhöhten Blutsenkungsreaktion (30 mm/h) bei normalem Differentialblutbild ohne Erhöhung der Leukozytenzahl. Elektrolyte, Nieren- und Leberwerte waren unauffällig. Ein CT von Hals, Thorax und Abdomen lieferte keine Hinweise auf einen Infektfokus oder eine Neoplasie. Erneut fanden sich serologisch keine Anhaltspunkte für eine infektiöse oder autoimmune Systemerkrankung (ANA, ANCA, Rheumafaktoren, Hepatitis- und HIV-Serologie negativ), ein familiäres Mittelmeerfieber konnte mittels Genanalyse ausgeschlossen werden. Die Immunglobuline im Serum, inklusive IgG-Subklassen, waren im Normbereich. Aufgrund eines positiven Eisbeutel-Tests (Besserung der subjektiv empfundenen und objektiv sehr diskret feststellbaren Ptose nach Auflegen eines Eisbeutels) wurde eine Myasthenia gravis in Betracht gezogen, dieser Verdacht bestätigte sich aber bei negativen Autoantikörpern gegen den Acetylcholin-Rezeptor nicht. Da die Ursache des systemischen Entzündungszustandes unklar blieb und die diskrete Ptose das einzige lokalisierende Symptom darstellte, erfolgte eine Magnetresonanztomographie (MRT) des Schädels, in der ein Pseudotumor der Glandula lacrimalis links auffiel (Abb. 1). Eine Biopsie dieses Befundes wurde veranlasst.
Abbildung 1: Magnetresonanztomographie des Schädels, Sagittalschnitt: Nachweis eines Pseudotumors der Glandula lacrimalis links (Pfeil).
Diagnose
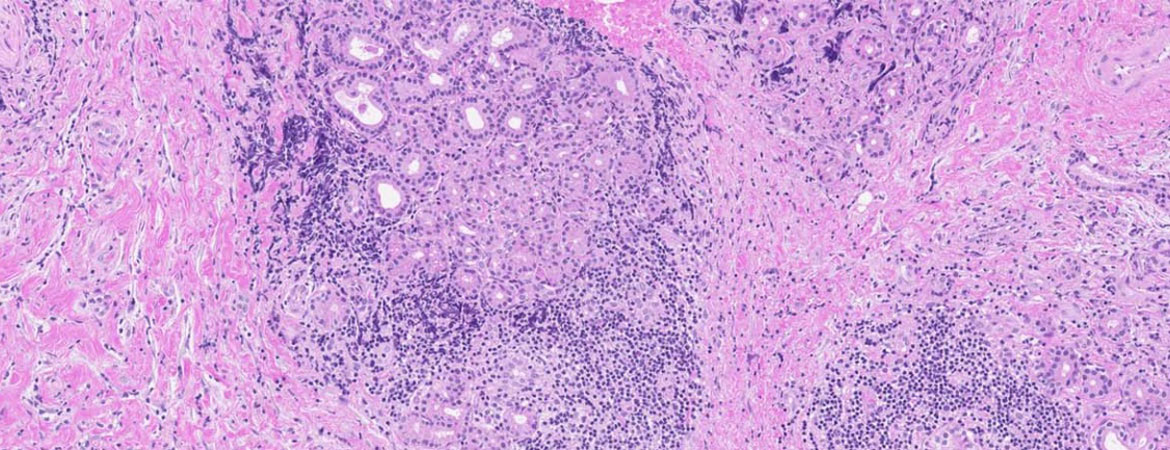
In der Histologie der Glandula lacrimalis zeigte sich ein plasmazellreiches Infiltrat, immunhistochemisch färbten sich Anteile der Plasmazellen positiv für IgG4. Anhaltspunkte für ein Lymphom oder eine Sarkoidose – als typische Differentialdiagnosen eines Pseudotumors der Glandula lacrimalis – fanden sich nicht (Abb. 2).
Abbildung 2: Gewebeprobe der Glandula lacrimalis. A) Übersicht (HE-Färbung): Ausgeprägte atrophisierende Fibrose mit wenigen Restdrüsenläppchen und chronisch-plasmazellulärer Entzündung. B) IgG-Immunhistochemie bei ausgeprägter artefizieller Hintergrundfärbung: zahlreiche IgG-positive Plasmazellen in einem entzündlichen Herd. C) IgG4-Immunhistochemie mit Nachweis IgG4-positiver Plasmazellen (<20% der IgG-positiven Plasmazellen). «Scale bar» in allen Ausschnitten 250 μm.
Im klinischen Kontext und ohne bioptische Hinweise auf das Vorliegen einer anderen Entität ist diese Befundkonstellation gut vereinbar mit der Diagnose einer IgG4-assoziierten Erkrankung (englisch: «IgG4-related disease» [IgG4-RD]) mit isoliertem Befall der Tränendrüse und systemischen Entzündungszeichen, auch wenn die histologischen Diagnosekriterien (siehe unten: lympho-plasmazelluläres Infiltrat mit >40% Plasmazellen positiv für IgG4, storiforme Fibrose und Phlebitis) nicht vollständig erfüllt waren. Dies wahrscheinlich aufgrund des längeren Krankheitsverlaufes mit ausgeprägter Fibrosierung der Gewebeprobe und bereits vorgängig erfolgter Steroidtherapie.
Therapie und Verlauf
Bereits die intraoperative Gabe von 250 mg Methylprednisolon, die zur Abschwellung erfolgt war, führte zu einer deutlichen Besserung sowohl des Allgemeinzustands des Patienten als auch der Entzündungswerte. Die anschliessende orale systemische Steroidtherapie (initiale Dosierung 0,5 mg/kg Körpergewicht [KG]) führte rasch zur vollständigen Remission der Beschwerden, sie konnte in der Folge über sechs Wochen problemlos ausgeschlichen werden. Der Patient blieb in den weiteren Kontrollen asymptomatisch.
Diskussion
Die IgG4-RD wird seit 2003 als eigenständige Entität beschrieben. Die Erkrankung ist histopathologisch charakterisiert durch eine lympho-plasmazelluläre Infiltration mit storiformer Fibrose und obliterativer Phlebitis, die unterschiedlichste Organe betreffen kann. Die polyklonalen Plasmazellen produzieren IgG4 und lassen sich entsprechend in einer immunhistochemischen Färbung darstellen. Während sich eine IgG4-RD als Multiorganerkrankung manifestieren kann, ist oft nur ein einzelnes Organ betroffen und eine Vielzahl vormals eigenständiger Erkrankungen wurde als Manifestationen einer IgG4-RD erkannt respektive wird heute unter dieser Entität subsummiert: Morbus Ormond (retroperitoneale Fibrose), Autoimmun-Pankreatitis, Küttner-Tumor (Sialadenitis), Mikulicz-Syndrom (Pseudotumor der Tränen- und Speicheldrüsen), Riedel-Struma, IgG4-assoziierte interstitielle Nephritis. Bei idiopathischen orbitalen Entzündungsprozessen wird ebenfalls häufig eine IgG4-RD als einzige Manifestation der Erkrankung gefunden («IgG4-related orbital disease» [IgG4-ROD]) [1].
Die IgG4-RD betrifft überwiegend Männer mit einem Durchschnittsalter von 67 Jahren [2].
Die Diagnose einer IgG4-RD wird histologisch gestellt. Während die Differentialdiagnose vor der Biopsie breit ist, macht die Suche nach anderen Entitäten wie Sarkoidose, Lymphom etc. nach eindeutigem Nachweis einer IgG4-RD im Gewebe mit Erfüllung aller histologischen Kriterien (histopathologisches Muster, entsprechende immunhistochemische Färbungen) wenig Sinn. Da – wie in unserem Fall – die histologischen Merkmale nicht immer eindeutig sind, muss die Diagnose aber immer im klinischen Kontext gestellt werden.
Eine erhöhte IgG4-Konzentration im Blut kann zwar zusammen mit einer typischen Klinik respektive einem bildgebenden Befund einen starken Hinweis auf eine IgG4-RD liefern, sie ist aber von begrenzter Sensitivität und Spezifität. Während praktisch alle Patienten mit einer IgG4-assoziierten interstitiellen Nephritis einen erhöhten IgG4-Spiegel im peripheren Blut zeigen, ist dies nur bei 70– 80% der Patienten mit histologisch gesicherter IgG4-assoziierter Autoimmun-Pankreatitis der Fall. Bei anderen extrapankreatischen Manifestationen einer IgG4-RD ist die diagnostische Wertigkeit des peripheren IgG4-Spiegels noch geringer [3]. Auch bei unserem Patienten fand sich eine normale Konzentration aller IgG-Subklassen im Blut. Umgekehrt können erhöhte IgG4-Konzentrationen im Blut auch bei gesunden Probanden beziehungsweise bei Patienten ohne IgG4-RD vorkommen und sollten nicht dazu verleiten, bei gesicherter anderer Diagnose (z.B. Sarkoidose) weitere Abklärungsschritte hinsichtlich einer IgG4-RD zu veranlassen. [4]
Meist verläuft die Krankheit subakut und jahrelang unerkannt; die meisten Patienten sind nicht schwerwiegend erkrankt [5]. Es kann zu temporären spontanen Remissionen kommen, Rezidive sind aber häufig [6]. Neben Symptomen aufgrund des spezifischen Organbefalls können auch unspezifische Allgemeinsymptome auftreten. Gewichtsverlust und generelle Abgeschlagenheit werden als häufigste Begleitsymptome beschrieben. Ein systemischer Entzündungszustand mit Fieber und CRP-Erhöhung, der bei unserem Patienten die Hauptmanifestation darstellte, findet sich nur bei einer Minderheit der Patienten (<10%) [3].
Therapeutisch spricht eine IgG4-RD sehr gut auf systemische Steroide an. Meistens können die Steroide über 4–6 Wochen ausgeschlichen werden. Gelingt dies nicht, kommen steroidsparende Medikamente wie Mycophenolat, Azathioprin oder Methotrexat zum Einsatz. In einzelnen therapieresistenten Fällen zeigte eine immunsuppressive Therapie mit Rituximab gute Effekte.
Schlussfolgerung
Die IgG4-RD stellt eine erst relativ kürzlich beschriebene Entität dar, findet aber seit ihrer Beschreibung zunehmend Beachtung und wird immer häufiger diagnostiziert. Je nach betroffenen Organen präsentiert sich eine vielfältige Klinik. Unser Patient mit im Vordergrund stehenden systemischen Entzündungszeichen zeigte eine sehr seltene Hauptmanifestation dieser Erkrankung. Es gibt bis anhin nur wenige beschriebene Fälle von IgG4-RD mit Fieber und erhöhten Entzündungszeichen im Sinne eines «fever of unknown origin» (FUO) [7].
Die sichere Diagnose einer IgG4-RD gelingt nur mittels Histologie, da die Sensitivität und Spezifität eines erhöhten IgG4-Spiegels im peripheren Blut begrenzt ist. Entscheidend war bei unserem Patienten mit unklarem Entzündungszustand, dass die Ptose trotz sehr diskreter und kaum objektivierbarer Klinik als einziges lokalisierendes Symptom erkannt und letztlich eine Gewebsdiagnose erzwungen wurde. Nur die sehr genaue Anamnese mit Fremdanamnese und klinischer Untersuchung hat uns schliesslich auf die richtige Spur gebracht.
Zusammenfassend zeigt unser Fallbericht, dass eine IgG4-RD als Ursache eines FUO respektive eines unklaren Entzündungszustandes in Betracht gezogen werden muss. Die anamnestischen und klinischen Hinweise dazu können allerdings diskret sein.
Das Wichtigste für die Praxis
• Eine IgG4-assoziierte Erkrankung («IgG4-related disease» [IgG4-RD]) kann sich als «fever of unknown origin» (FUO) mit nur sehr diskreten lokalisierenden Symptomen manifestieren.
• Zur sicheren Diagnose einer IgG4-RD ist eine Histologie unabdingbar; die IgG4-Konzentration im peripheren Blut ist von geringer Sensitivität und Spezifität.
• Die Erkrankung spricht ausgezeichnet auf Steroide an, Rezidive sind aber möglich. Entsprechend ist ein engmaschiges Follow-up nötig.
Disclosure statement
Die Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Correspondance
KD Dr. med. Elisabeth Weber Departement Innere Medizin und Spezialdisziplinen Stadtspital Triemli Zürich Birmensdorferstr. 497 CH-8063 Zürich elisabeth.weber[at] triemli.zuerich.ch
Literatur
1 Andron A, Hostovsky A, Nair AG et al. The impact of IgG-4 ROD on the diagnosis of orbital tumors: A retrospective analysis. Orbit. 2017;36(6):359–64.
2 Kamisawa T, Zen Y, Pillai S et al. IgG4-related disease. Lancet. 2015;385:1460–71.
3 Mahajan VS, Mattoo H, Deshpande V et al. IgG4-related disease. Annu Rev Pathol Mech Dis. 2014;9:315–47.
4 Carruthers MN, Khosroshahi A, Augustin T et al. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2015;74:14–8.
5 Stone JH, Zen Y, Deshpande V. IgG4-Related Disease. N Engl J Med. 2012;366:539–51.
6 Hamano H, Tanaka E, Ishizaka N et al. IgG4 related Disease – A systemic Disease that Deserves Attention Regardless of one’s Subspecialty. Intern Med. 2018;57(9):1201–7.
7 Sebastian A, Sebastian M, Missterska-Skora M et al. The variety of clinical presentations in IgG4-related disease in Rheumatology. Rheumatol Int. 2018;38:303–9.