Un patient se présente au service des urgences en raison d’une parésie de la jambe, d’une rétention urinaire, d’un épisode de hoquet de deux jours et d’un épisode de vision floue de plusieurs heures.
Contexte
Nous rapportons un cas de neuromyélite optique (NMO), une maladie rare du système nerveux central (SNC). Elle a causé une hyponatrémie avec constellation de syndrome de sécrétion inappropriée d’hormone antidiurétique (SIADH), déclenché par l’atteinte inflammatoire de l’hypothalamus.
Présentation du cas
Anamnèse
Un patient de 67 ans s’est présenté au service des urgences en raison d’une parésie de la jambe gauche nouvellement survenue et d’une rétention urinaire. Hormis une hypertension artérielle, le patient ne présentait pas d’antécédents médicaux. Lors de l’anamnèse systémique, il a rapporté un épisode de hoquet de deux jours et un épisode de vision floue de plusieurs heures ces jours derniers. Déjà dix jours auparavant, le patient avait été hospitalisé en raison d’une faiblesse physique prononcée avec inappétence et humeur dépressive. Les symptômes avaient été attribués à une hyponatrémie hypotonique modérée, compatible avec un SIADH. Sur le plan étiologique, l’hyponatrémie avait été interprétée en tant qu’effet indésirable du traitement antihypertenseur par inhibiteur de l’enzyme de conversion de l’angiotensine, avec en parallèle une consommation accrue d’alcool. Elle s’était normalisée durant l’hospitalisation moyennant une restriction des apports liquidiens et une interruption temporaire du traitement antihypertenseur. Il a été jugé que les troubles de la sensibilité (sensation de brûlure) dans la région de la cuisse droite et de l’épaule gauche, déjà présents à ce moment-là, s’expliquaient le plus probablement par le déséquilibre électrolytique. Une étiologie en lien avec une toxicité alcoolique avait également été envisagée dans le diagnostic différentiel. Le patient ne s’était pas plaint d’autres symptômes d’une maladie cérébrale primaire avec atteinte de l’hypothalamus, tels que des troubles de la sensation de faim ou du rythme sommeil-veille.
Statut
A son admission dans notre hôpital, le patient était parfaitement orienté, afébrile et euvolémique, avec un statut cardiopulmonaire et abdominal normal. L’examen neurologique clinique était évocateur d’une myélopathie, avec des éléments d’un syndrome de Brown-Séquard. Le patient présentait une forte parésie de la jambe gauche avec réflexes ostéo-tendineux abolis du côté gauche et signe de Babinski positif des deux côtés. Il existait un trouble sensitif dissocié sous la Th4 au sens d’une hypoesthésie et hypoalgésie du côté droit. Le tonus sphinctérien était affaibli, et le patient ne pouvait se tenir debout qu’avec beaucoup d’aide. Le statut des nerfs cérébraux était normal.
Diagnostic
A l’admission, les analyses de laboratoire ont à nouveau montré une hyponatrémie hypotonique et une constellation de SIADH avec une urine hypertonique. Les autres valeurs étaient normales (tab. 1). Une hypothyroïdie en tant que cause de l’hyponatrémie euvolémique a été exclue sur la base d’une TSH normale, et un dosage de la cortisone à jeun visant à exclure une insuffisance corticosurrénale n’a malheureusement pas été réalisé.
Tableau 1: Valeurs de laboratoire lors de la première présentation chez le médecin.
Sérum
Valeurs de référence
Sodium
125 mmol/l
136–145 mmol/l
Potassium
4,3 mmol/l
3,5–5,1 mmol/l
Créatinine
70 μmol/l
64–111 μmol/l
Urée
3,8 mmol/l
3,0–9,2 mmol/l
Osmolarité
265 mosmol/kg
280–300 mosmol/kg
TSH
0,62 mU/l
0,35–4,94 mU/l
Urine
Sodium
69 mmol/l
Potassium
15 mmol/l
Osmolarité
277 mosmol/kg
TSH: thyréostimuline
Un SIADH paranéoplasique dû à un carcinome bronchique ou une autre maladie pulmonaire avait déjà été exclu par tomodensitométrie thoracique lors de la précédente hospitalisation. Ni l’examen clinique ni l’anamnèse n’ont amené à suspecter une autre maladie maligne. Une étiologie médicamenteuse du SIADH a été exclue sur la base de l’anamnèse, et aucun indice de causes plus rares du SIADH n’a été trouvé. Le diagnostic de suspicion d’une maladie neurologique primaire a finalement prédominé. Nous avons réalisé une imagerie par résonance magnétique (IRM) rachidienne et cérébrale, qui a montré de multiples lésions intramédullaires extensives en T2 avec absorption non homogène du produit de contraste dans la moelle (fig. 1). Au niveau cérébral, de multiples lésions inflammatoires supratentorielles ont été visualisées, notamment dans la substance blanche, au niveau périventriculaire, et dans l’hypothalamus proximal (fig. 2). Une atteinte des nerfs optiques n’a pas pu être visualisée à l’IRM, mais a ensuite été confirmée par un retard de conduction bilatéral à la mesure des potentiels évoqués visuels.
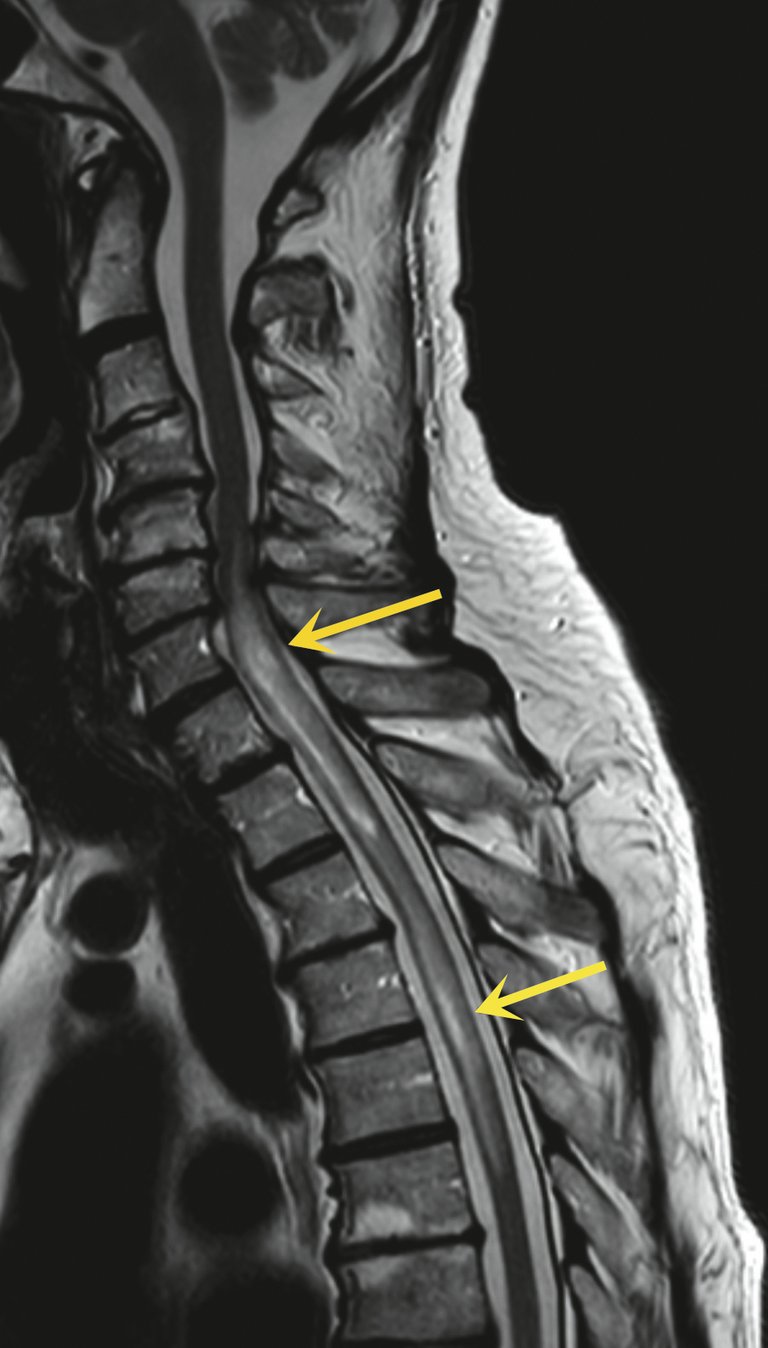
Figure 1: IRM en coupe sagittale, séquence T2 «turbo spin echo» (TSE), montrant deux longues zones intramédullaires hyperintenses (flèches jaunes), avec un large oedème périfocal dans la moelle cervicale inférieure et la moelle thoracique supérieure.
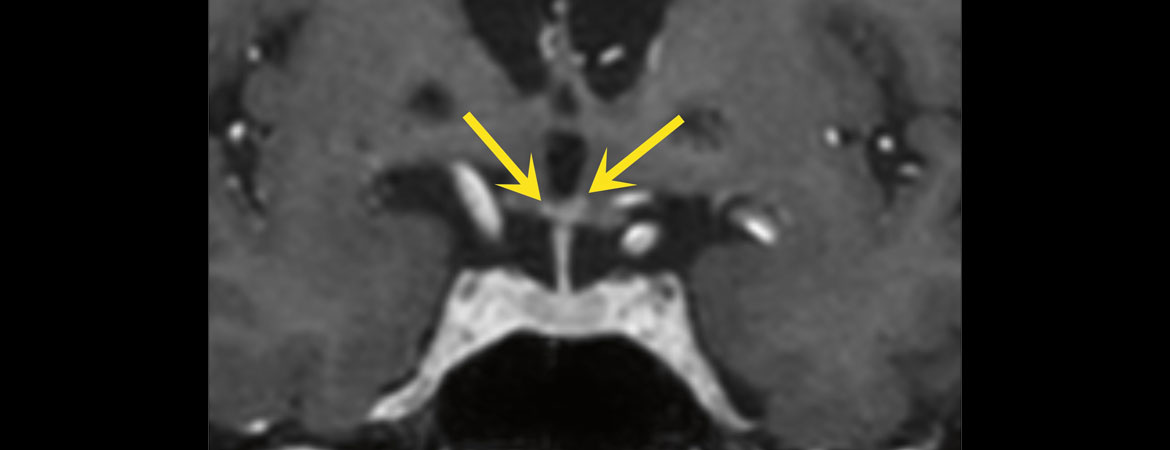
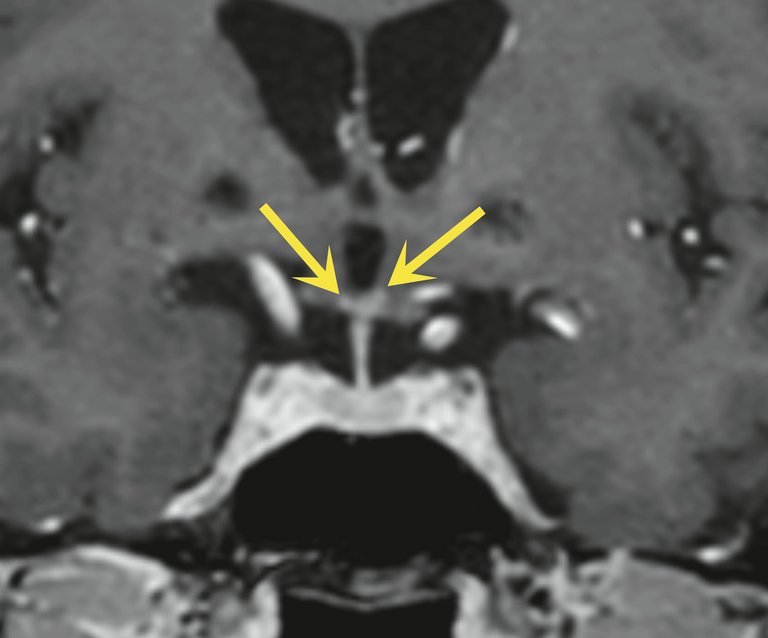
Figure 2: IRM en coupe coronale, séquence T1 «turbo field echo» (TFE) m-Dixon, après administration de produit de contraste montrant de petites prises de contraste inflammatoires punctiformes bilatérales dans l’hypothalamus (flèches jaunes).
En présence de démyélinisations centrales et d’une atteinte des nerfs optiques, nous avons pensé à une sclérose en plaques (SEP). Toutefois, les démyélinisations extensives à l’IRM ne concordaient pas, et seule une faible concentration de bandes oligoclonales a été mise en évidence dans le liquide céphalo-rachidien (LCR). La ponction lombaire ayant révélé une pléïocytose lymphocytaire de 50 cellules/μl et une perturbation importante de la barrière hémato-encéphalique, une cause infectieuse était aussi envisageable dans le diagnostic différentiel. Cependant, aucun élément allant en ce sens n’a été trouvé: glucose et lactate normaux à la ponction lombaire, mise en culture bactérienne du LCR négative et sérologies négatives (syphilis, borrélies, VIH, VHS, VVZ et VHH-6). Une vascularite cérébrale semblait peu probable en présence d’un test de dépistage par auto-anticorps négatif (anticorps antinucléaires, anticorps anti-ADNdb, anticorps anti-SSB/La, anticorps anti-SSA/Ro, complément C3/C4 négatifs).
Au regard de la myélite longitudinale étendue et de la neuropathie optique bilatérale, nous avons finalement posé le diagnostic de NMO. De façon concordante, les anticorps anti-aquaporine-4 (Ac anti-AQP4) sériques étaient hautement positifs (1:320, norme <1:10).
Traitement et évolution
La corticothérapie de choc initiale, avec 1 g de méthylprednisolone par jour pendant sept jours, et la corticothérapie d’entretien consécutive par voie orale n’ont pas apporté d’amélioration neurologique. Nous avons donc procédé à une plasmaphérèse. Malheureusement, un trouble de la coagulation avec formation d’un hématome extensif anémiant sur la jambe parétique gauche est survenu en tant qu’effet indésirable et nous avons dû interrompre la plasmaphérèse précocement après cinq cycles. A ce moment-là, une légère amélioration de la force de la jambe gauche s’était manifestée, mais le patient se déplaçait toujours en fauteuil roulant. Concernant l’hyponatrémie modérée initiale, une solution de chlorure de sodium hyperosmolaire avait été perfusée pendant une courte durée à l’admission et déjà arrêtée après quelques heures en raison d’une élévation du sodium dans la norme inférieure. Par la suite, le sodium sérique et l’osmolarité sérique sont restés dans la norme inférieure.
Près de quatre semaines après l’admission à l’hôpital, nous avons pour la première fois administré 1 g de rituximab pour prévenir les récidives. Suite à une neuroréhabilitation de quatre mois, le patient était mobile sans moyens auxiliaires. Une légère ataxie résiduelle marquée à gauche était présente. Le traitement par rituximab sera continué à intervalles de six mois pendant les deux prochaines années.
Discussion
La NMO est une maladie inflammatoire auto-immune du SNC atteignant typiquement les nerfs optiques et la moelle épinière. Cliniquement, la NMO se manifeste par des troubles de la vision, des parésies, des troubles de la sensibilité et une dysfonction vésicale. En cas d’atteinte du tronc cérébral, et notamment de l’area postrema, des nausées, un hoquet et une insuffisance respiratoire peuvent survenir [1].
La majorité des patients présentent une évolution progressive de la maladie avec des poussées répétées. Des évolutions monophasiques ont plus rarement été décrites. Au total, les maladies du spectre de la NMO touchent en moyenne 2/100 000 personnes. Les femmes sont plus touchées que les hommes, et les populations non caucasiennes sont davantage touchées que la population caucasienne.
La maladie a initialement été décrite en tant que syndrome optico-spinal avec névrite optique et myélite transverse simultanées par Eugene Devic (syndrome de Devic) en 1894. La distinction, notamment avec la SEP, est longtemps restée floue. C’est uniquement avec le développement des procédés d’imagerie et des analyses du LCR que des différences croissantes ont émergé. Contrairement à la SEP, en cas de NMO, des bandes oligoclonales ne sont en principe pas mises en évidence dans le LCR, il est plus fréquent que l’IRM cérébrale soit normale, et les démyélinisations de la moelle épinière sont plus étendues. Si des démyélinisations cérébrales surviennent tout de même dans la NMO, elles sont typiquement localisées le long du 3e et du 4e ventricule latéral. Ce n’est qu’avec la découverte en 2004 de l’auto-anticorps sérique hautement spécifique dirigé contre le canal de l’aquaporine-4 (IgG anti-AQP4) que la NMO a été consolidée en tant qu’entité propre. Toutefois, environ 30% de tous les patients atteints de NMO sont négatifs pour les IgG anti-AQP4, et un anticorps dirigé contre la myéline oligodendrocytaire (IgG anti-MOG) peut être mis en évidence chez environ un quart d’entre eux [2]. En 2007, le terme «NMO spectrum disorder» (NMOSD) a été créé, regroupant à la fois la NMO classique et les formes incomplètes. En 2015, les critères pour la pose du diagnostic de NMOSD, qui sont aujourd’hui en vigueur, ont été établis [3].
Les mécanismes précis de survenue de la maladie ne sont pas élucidés. L’Ac anti-AQP4 lui-même semble jouer un rôle central dans la pathogenèse, car en se liant aux canaux astrocytaires dans le SNC, il contribue directement à l’inflammation, à la démyélinisation et à la lyse des cellules cibles médiée par le complément.
Les poussées aiguës sont traitées par une corticothérapie de choc. En l’absence de réponse, une plasmaphérèse peut être envisagée. Pour prévenir les récidives, une immunosuppression est recommandée pendant au moins cinq ans. Les stéroïdes sont souvent associés à de l’azathioprine, du mycophénolate mofétil ou du rituximab, mais il n’existe aucune comparaison directe dans des études prospectives. Fait intéressant, les traitements immunomodulateurs classiques contre la SEP sont inefficaces voire potentiellement néfastes. Le pronostic de la NMO est plus mauvais que celui de la SEP, les poussées sont souvent plus sévères et le rétablissement neurologique est moins bon.
Dans la littérature, les hyponatrémies touchent environ 15% de tous les patients atteints de NMOSD et un SIADH est présent chez la plupart d’entre eux, comme chez notre patient [4]. De potentiels mécanismes du SIADH dans les NMOSD sont les affections immunologiques de l’hypothalamus riche en AQP4, avec sortie d’ADH, ou de l’organe circumventriculaire, également impliqué dans la régulation de l’équilibre sodique. Etant donné que les NMOSD, contrairement à la SEP, affectent préférentiellement l’hypothalamus, le SIADH survient plus souvent que dans la SEP. Une cause bien plus rare d’hyponatrémie dans de nombreuses maladies neurologiques cérébrales est le syndrome de perte de sel d’origine cérébrale. Ce dernier ne se différencie guère du SIADH au niveau des analyses de laboratoire, mais il s’accompagne d’une déshydratation clinique due à une polyurie induite par une excrétion rénale de sodium excessive.
L’essentiel pour la pratique
• Lors du diagnostic différentiel des hyponatrémies, surtout en cas de constellation de SIADH, il convient de penser à des maladies neurologiques.
• En cas de névrite optique et de myélite longitudinale étendue, il convient de songer à une neuromyélite optique (NMO). Les anticorps IgG anti-AQP4 sont hautement spécifiques, mais il existe également des NMOSD avec des anticorps IgG anti-AQP4 négatifs.
• Lors de la poussée aiguë, la NMO est traitée avec une corticothérapie de choc, et par plasmaphérèse dans les cas graves. Une immunosuppression est mise en œuvre pour prévenir les récidives.
• La distinction avec la sclérose en plaques (SEP) est centrale sur le plan thérapeutique, car les médicaments anti-SEP classiques peuvent potentiellement aggraver la NMO.
Disclosure statement
Les auteurs n’ont pas déclaré des obligations financières ou personnelles en rapport avec l’article soumis.
1 Weinshenker BG, Wingerchuk DM. Neuromyelitis Spectrum Disorders. Mayo Clin Proc. 2017;92(4):663–79.
2 Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Luccinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–12.
3 Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177–89.
4 Jin S, Long Z, Wang W, Jiang B. Hyponatremia in neuromyelitis optica spectrum disorders: Literature review. Acta Neurol Scand. 2018;138(1):4–11.