a Klinik für Pneumologie, Universitätsspital Zürich, Zürich; b Pneumologie, Medizinische Klinik, Zuger Kantonsspital, Baar; c Pneumologie, Departement Innere Medizin, Stadtspital Zürich, Standort Waid, Zürich
Un patient de 35 ans a été admis aux urgences en raison d’une aggravation de son état malgré un traitement pour une suspicion de pneumonie COVID-19 avec surinfection bactérienne.
Contexte
Même en période de pandémie de coronavirus, il est important de maintenir ouverte la palette des diagnostics différentiels et, en particulier en présence de tests SARS-CoV-2 négatifs répétés, de compléter de manière ciblée ce diagnostic suspecté en approfondissant l’anamnèse et les examens afin de ne pas négliger des tableaux cliniques rares, potentiellement dangereux, tels qu’une vascularite.
Rapport de cas
Anamnèse
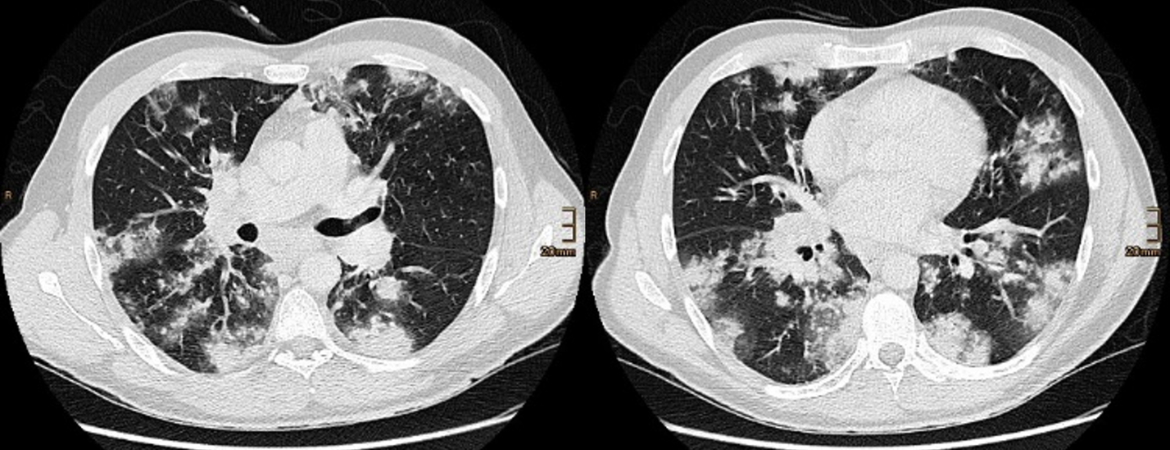
Un patient âgé de 35 ans, souffrant d’un asthme bronchique connu, de polypose nasale et de diabète sucré de type 1 depuis l’âge de 11 ans, a consulté son médecin de famille en raison de douleurs thoraciques lancinantes à gauche ainsi qu’une dyspnée d’effort et une sensation de froid. En présence d’une diminution de la transparence à la radiographie thoracique, d’une leucocytose (hémogramme réalisé sans différenciation) et d’un taux accru de protéine C-réactive (CPR), un traitement antibiotique par co-amoxicilline a été débuté en ambulatoire. Le test de COVID-19 était négatif. Après cinq jours, le patient a dû être hospitalisé en raison de fièvre, d’une dégradation de son état général et d’une désaturation en oxygène (SpO2 89%). Sur le plan radiologique, la tomodensitométrie (TDM) du thorax a révélé des infiltrats consolidés à répartition bilatérale symétrique avec des opacités environnantes en verre dépoli ainsi qu’une lymphadénopathie médiastinale et hilaire (fig. 1).
Figure 1: Tomodensitométrie thoracique (coupes axiales) lors de la première consultation à l’hôpital. Consolidations à répartition bilatérale avec opacités en verre dépoli.
Malgré un frottis de COVID-19 toujours négatif, le résultat était compatible avec un stade consolidé et une superinfection bactérienne liée à une pneumonie de COVID-19. La recherche d’agents pathogènes typiques et atypiques est restée infructueuse. A la suite de l’escalade du traitement antibiotique à la ceftriaxone et la clarithromycine et de l’ajout de stéroïdes oraux, le patient a pu être déchargé au bout de trois jours. Après achèvement du traitement, l’état du patient s’est toutefois à nouveau dégradé de sorte que le médecin de famille a dû l’adresser à un autre hôpital.
Examen clinique et résultats
Au service des urgences, le patient était tachypnéique (fréquence respiratoire 28/min), tachycarde (fréquence cardiaque 135/min), normotendu (pression artérielle 132/75 mm Hg) et légèrement subfébrile (température 37,5 °C). La saturation en oxygène était de 95% à l’air ambiant. L’auscultation a permis de déceler une sibilance ubiquitaire en fin d’expiration ainsi que des râles crépitants bibasaux. Les bruits cardiaques étaient rythmiques et tachycardes, le patient ne présentait pas d’œdème de jambe, ni de turgescence jugulaire, le réflexe hépato-jugulaire était négatif. Le reste du statut était normal.
L’hémogramme a révélé une éosinophilie prononcée à raison de 4,43 G/l (valeur normale 0–0,7 G/l), correspondant à 19,6% du nombre total de globules blancs, avec une légère leucocytose et un nombre normal de plaquettes. Les analyses biochimiques ont indiqué un taux accru de CRP de 48 mg/l (valeur normale <5 mg/l) pour un taux normal de procalcitonine, des biomarqueurs cardiaques accrus avec un taux de troponine T hautement sensible (hs) 798 ng/l (valeur normale <14 ng/l), un taux de créatine kinase (CK) de 257 U/l (valeur normale <190 U/l) et un taux de «N-terminal pro brain natriuretic peptide» (NT-proBNP) de 3551 ng/l (valeur normale <85,8 ng/l). Les valeurs rénales et hépatiques étaient normales, tout comme les électrolytes. Le sédiment urinaire était également normal. Le test de réaction de polymérisation en chaîne (PCR) au SARS-CoV-2 était à nouveau négatif.
La TDM thoracique présentait toujours les consolidations bilatérales avec opacités en verre dépoli. La TDM des sinus a révélé une pansinusite. L’électrocardiographie (ECG) a permis d’observer un rythme sinusal tachycarde avec élévations du segment ST en dérivations I et aVL ainsi qu’une élévation minimale en V5 et V6, en l’absence de troubles de dépolarisation ou repolarisation. L’échocardiographie transthoracique a révélé une fraction d’éjection ventriculaire gauche légèrement réduite (49%) en présence d’un épaississement de la paroi d’aspect œdémateux étendu avec une hypokinésie inféro- et antérolatérale entre la zone basale et la zone apicale et atteignant aussi l’aire septale – en présence de cavités cardiaques de taille normale et sans indication d’une hypertension pulmonaire.
Pour résumer, il existait une éosinophilie massive, de multiples consolidations pulmonaires, des biomarqueurs cardiaques accrus, un asthme bronchique connu et une polypose nasale avec pansinusite à la TDM, permettant d’établir la suspicion élevée d’une granulomatose éosinophilique avec polyangéite (GEPA, anciennement syndrome de Churg et Strauss) avec atteinte cardiaque.
Traitement et évolution
En raison de l’atteinte cardiaque, le patient a été transféré dans un centre afin de réaliser une coronarographie au vu des élévations du ST et en présence de diabète sucré de type 1 comme facteur de risque cardiovasculaire. Celle-ci a montré des coronaires exemptes de sténoses et une fonction ventriculaire gauche normale à la ventriculographie. Sur la base de ce résultat, les biomarqueurs cardiaques accrus ont été interprétés comme une atteinte cardiaque en présence d’une granulomatose éosinophilique avec polyangéite. Les valeurs rhumatologiques complémentaires de laboratoire ont révélé un taux d’immunoglobuline E (IgE) légèrement accru, mais des anticorps anti-cytoplasme des polynucléaires neutrophiles (ANCA) négatifs ainsi que des valeurs normales d’anticorps anti-myéloperoxydase (anti-MPO) et anti-protéinase 3 (anti-PR3). Les anticorps anti-nucléaires (ANA) étaient également négatifs.
Au vu de l’atteinte cardiaque sévère, 1 g de méthylprednisolone a immédiatement été administré en intraveineuse et le traitement poursuivi à cette dose pendant trois jours – en présence d’une tachycardie persistante sous surveillance dans la «Intermediate Care Unit». Sous le traitement, les granulocytes éosinophiliques ont retrouvé une valeur normale dans un délai de douze heures (de 4,43 à 0,47 G/l; tab. 1). Le patient est resté en compensation cardiaque sans traitement diurétique et son état s’est rapidement amélioré.
Tableau 1: Evolution des paramètres de laboratoire sous traitement
Jour 1
Jour 2
Jour 4
Jour 6
Jour 7
Jour 8
Jour 9
Jour 30
3 mois
Eosinophiles (valeur normale: 0–0,7G/l, 0–7%)
4,43 G/l (19,6%)
0,47 G/l (3,6%)
0,02 G/l (0,1%)
1,11 G/l (7,6%)
0,49 G/l (3,9%)
0,29 G/l (2,4%)
0,47 G/l (4%)
0,01 G/L
0,09 G/L
Troponine T hs (valeur normale <14ng/l)
798 ng/l
456 ng/l
665 ng/l
810 ng/l
491 ng/l
381 ng/l
309 ng/l
12 ng/l
12 ng/l
CRP (valeur normale < 5 mg/l)
48 mg/l
38 mg/l
13 mg/l
4,2 mg/l
2,8 mg/l
2,1 mg/l
1,4 mg/l
<0,6 mg/l
0,8 mg/l
Traitement
Début méthylprednisolone 1 g
Méthylprednisolone 1 g
Prednisone 75 mg
Prednisone 75 mg
Prednisone 50 mg Cyclophosphamide 1 g
Prednisone 50 mg
Prednisone 50 mg
Prednisone 30 mg Cyclophosphamide (6 cycles)
Prednisone 20 mg Azathioprine 100 mg
hs: hautement sensible; CRP: protéine C-réactive.
Après réduction des stéroïdes et passage à 75 mg de prednisone à partir du quatrième jour d’hospitalisation, une nouvelle hausse des éosinophiles et de la troponine T hs est survenue. En même temps sont apparues des valeurs glycémiques accrues induites par stéroïdes en présence d’un diabète sucré de type 1 connu, de sorte que nous avons opté pour un traitement par cyclophosphamide avec cryoconservation préalable de spermatozoïdes. La première dose de cyclophosphamide a été administrée en stationnaire et bien tolérée. Ce traitement a été poursuivi en ambulatoire d’abord deux fois par semaine jusqu’à la troisième dose, puis toutes les trois semaines.
Sous ce traitement, les éosinophiles et les biomarqueurs cardiaques ont régressé. Lors de l’échocardiographie de suivi, nous avons objectivé une normalisation de la fraction d’éjection ventriculaire gauche et un rétablissement général des troubles moteurs régionaux de la paroi (à l’exception de la zone latérale) avec toutefois un œdème persistant au niveau de la paroi. L’imagerie par résonance magnétique (IRM) cardiaque réalisée deux semaines plus tard a montré une fonction systolique ventriculaire gauche normale supérieure (fraction d’éjection 76%), aucun trouble moteur régional de la paroi, mais un rehaussement médio-mural tardif au gadolinium avec œdème myocardique et, ainsi, des signes d’inflammation au niveau antéro-latéral basal jusqu’à médio-ventriculaire et latéro-apical.
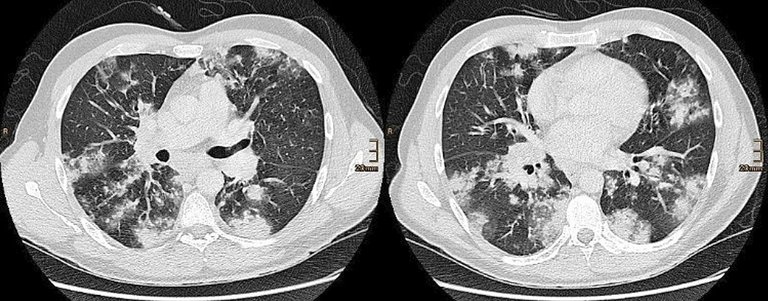
Après au total six administrations de cyclophosphamide et une réduction concomitante de prednisone à 20 mg/d, plus aucune modification inflammatoire n’était visible lors d’une nouvelle IRM cardiaque trois mois plus tard, en présence d’un taux de troponine T hs toujours normal. A la TDM, les modifications pulmonaires avaient presque complètement disparu (fig. 2).
Dans une prochaine étape, un traitement par azathioprine a été débuté pour épargner les stéroïdes.
Figure 2: Tomodensitométrie thoracique (coupes axiales) deux mois après le début du traitement. Parenchyme pulmonaire avec résultat quasiment normal.
Discussion
La GEPA est une angéite granulomateuse allergique principalement des petits vaisseaux, qui s’accompagne de granulomes extravasculaires et d’une hyperéosinophilie systémique [1]. L’étiologie et la pathogenèse exacte sont inconnues. Il s’agit typiquement d’une éosinophilie due à une apoptose réduite et un trouble de la réponse immunitaire ainsi qu’une fonction anormale des granulocytes éosinophiles.
Le tableau clinique présente trois phases: La phase prodromique s’exprime sous forme de maladie atopique, notamment une rhinite allergique ou un asthme bronchique. Ensuite se développe la phase éosinophilique présentant une éosinophilie sanguine et une invasion d’éosinophiles dans plusieurs organes, en particulier les poumons. Enfin survient la phase vascularitique, caractérisée par une vascularite systémique potentiellement mortelle des vaisseaux de moyen et petit calibre, souvent liée à une granulomatose vasculaire et extravasculaire. La phase vascularitique apparaît en moyenne douze ans après le diagnostic de l’asthme [2, 3].
Selon les critères de classification de l’«American College of Rheumatology» (ACR), le diagnostic repose sur la mise en évidence des résultats suivants [4, 5]:
Lorsqu’au moins quatre des six critères sont remplis, le diagnostic peut être établi avec une forte probabilité (spécificité 99%, sensibilité 85%), la confirmation du diagnostic au moyen de l’histologie n’est alors plus requise [4].
Au laboratoire, 30–60% des personnes concernées présentent des ANCA positifs, ceux-ci sont spécifiques à la MPO dans 96% des cas et à la PR3 dans 4% des cas [2]. Sur le plan radiologique, il est possible de mettre en évidence des consolidations périphériques à la radiographie thoracique conventionnelle. La TDM des poumons permet surtout d’observer des consolidations périphériques et multiples avec des opacités en verre dépoli. Il est moins fréquent de constater des nodules centro-lobulaires, épaississements de la paroi bronchique et ectasies bronchiques. L’hémorragie alvéolaire est très rare et survient principalement en cas de positivité des ANCA. La cavitation des lésions est atypique.
Le lavage broncho-alvéolaire présente typiquement une éosinophilie prononcée (environ 30%). Pour confirmer le diagnostic, les organes touchés peuvent être biopsiés, révélant des granulomes extravasculaires, une éosinophilie, une angéite nécrosante ainsi que des infiltrations éosinophiliques tissulaires. L’atteinte rénale doit être recherchée, mais elle est plus rare que pour les autres vascularites.
Une atteinte cardiaque survient dans près de 15–60% des cas de GEPA, en particulier chez les patientes et patients présentant des ANCA négatifs. Elle peut se manifester sous forme d’une myocardite, péricardite, arythmie, artérite des coronaires, valvulopathie et finalement une insuffisance cardiaque. Malgré l’absence clinique d’une symptomatique cardiaque et de modifications à l’ECG, le cœur peut être atteint dans 40% des cas. Une électrocardiographie initiale est ainsi indiquée dans tous les cas. En présence d’échocardiographie anormale, une IRM cardiaque doit être réalisée. Une coronarographie et une biopsie endomyocardique ne sont que rarement indiquées. Le traitement immédiat est essentiel, car une atteinte cardiaque cliniquement manifeste s’accompagne d’un mauvais pronostic et provoque 50% des cas de décès associés à la GEPA.
La GEPA est traitée par corticostéroïdes [6]. En fonction du degré de sévérité de la maladie, des médicaments immunosuppresseurs complémentaires, conventionnellement la cyclophosphamide, le méthotrexate ou l’azathioprine, sont prescrits. Dans une étude, le rituximab a entraîné une rémission et une réduction de la dose de corticostéroïdes chez près de la moitié des patientes et patients après douze mois [6]. Le mépolizumab (anticorps dirigé contre l’interleukine 5) est autorisé pour le traitement de la GEPA et semble avoir un effet épargnant les glucocorticostéroïdes [7].
En raison de la bonne réponse aux corticostéroïdes, le pronostic de la GEPA est favorable en cas d’atteinte pulmonaire isolée. Le pronostic est plus mauvais en cas d’atteinte cardiaque et gastrointestinale, de protéinurie >1g/d, d’insuffisance rénale ou d’atteinte du système nerveux central. Un traitement prolongé, généralement combiné, à base de cortisone et de médicaments épargnant les stéroïdes est alors requis [6].
Les diagnostics différentiels de la GEPA à envisager sont la «aspirin-exacerbated respiratory disease», une pneumonie chronique à éosinophiles, une aspergillose broncho-pulmonaire allergique, un syndrome d’hyperéosinophilie ou d’autres vascularites (granulomatose avec polyangéite, polyangéite microscopique) [3].
Les diagnostics différentiels des consolidations pulmonaires, comme présentes chez notre patient, incluent une pneumonie organisée cryptogénique (POC) ainsi que des infections bactériennes ou virales, principalement une pneumonie de COVID-19, bien que, dans ce dernier cas, la lymphadénopathie médiastinale serait atypique.
L’essentiel pour la pratique
Même en présence de symptômes respiratoires supposément typiques, d’une imagerie concordante et d’une probabilité a priori en période de pandémie, il convient d’envisager rapidement d’autres diagnostics différentiels, en particulier les vascularites, lorsque le test SARS-CoV-2 est négatif.
La granulomatose éosinophilique avec polyangéite (GEPA) est généralement une maladie systémique présentant un asthme bronchique, une éosinophilie et une rhinosinusite chronique, associés à une vascularite des artères de petit et moyen calibre.
Les critères de classification de l’«American College of Rheumatology» de 1990 peuvent être utilisés pour le diagnostic.
Un traitement corticostéroïde systémique hautement dosé, débuté immédiatement, peut éviter des états potentiellement mortels.
En cas d’atteinte cardiaque, une confirmation rapide du diagnostic et un début immédiat du traitement sont décisifs pour le pronostic.
Karzan Nasih Ali, médecin dilpômé
Klinik für Pneumologie,
Universtitätsspital Zürich, Zürich
Informed consent
Un consentement éclairé écrit est disponible pour la publication.
Correspondance
Karzan N. Ali
Klinik für Pneumologie
Universitätsspital Zürich
Rämistrasse 40
CH-8091 Zürich
Références
1 Hauser T, Mahr A, Metzler C, Coste J, Sommerstein R, Gross WL, et al. The leucotriene receptor antagonist montelukast and the risk of Churg-Strauss syndrome: a case-crossover study. Thorax. 2008;63(8):677–82.
2 Mouthon L, Dunogue B, Guillevin L. Diagnosis and classification of eosinophilic granulomatosis with polyangiitis (formerly named Churg-Strauss syndrome). J Autoimmun. 2014;48–49:99–103.
3 Cottin V, Bel E, Bottero P, Dalhoff K, Humbert M, Lazor R, et al. Respiratory manifestations of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Eur Respir J. 2016;48(5):1429–41.
4 Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
5 Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990;33(8):1094–100.
6 Groh M, Pagnoux C, Baldini C, Bel E, Bottero P, Cottin V, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur J Intern Med. 2015;26(7):545–53.
7 Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med. 2017;376(20):1921–32.