Un choc est une défaillance circulatoire avec un approvisionnement insuffisant en oxygène des organes. La partie 1 de cet articel de revue aborde les causes, la physiopathologie et la clinique des quatre formes de choc.
Introduction
Un choc est une défaillance circulatoire avec un déséquilibre entre les apports et les besoins en oxygène, qui peut exister au niveau macrocirculatoire, microcirculatoire et cellulaire. Un diagnostic précoce et un traitement ciblé sont essentiels afin d’éviter des dommages organiques irréversibles. Un traitement adéquat présuppose la connaissance des différentes formes de choc, de leurs causes et de leur physiopathologie. La première partie de cet article de revue aborde les mécanismes et la clinique des quatre formes de choc. La deuxième partie1 sera consacrée aux possibilités thérapeutiques.
Principes physiologiques
Macrocirculation
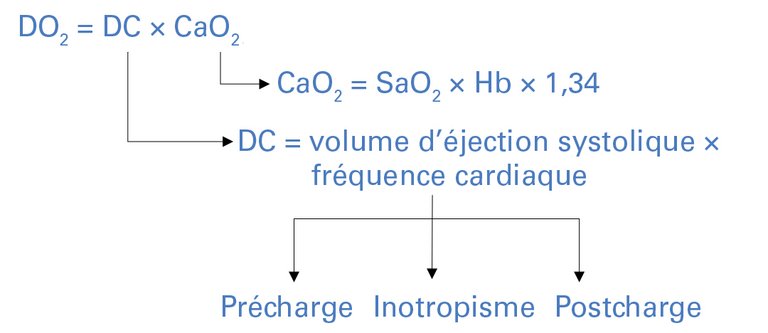
Le transport en oxygène (DO2) correspond à la quantité d’oxygène transportée des poumons vers la microcirculation. Les mécanismes de survenue des chocs et les approches thérapeutiques en découlent (fig. 1).
Figure 1: Transport en oxygène (DO 2 ) et ses facteurs d’influence.
CaO 2 : contenu artériel en oxygène (formule simplifiée sans oxygène dissous); DO 2 : transport en oxygène; Hb: hémoglobine (en g/dl); DC: débit cardiaque; SaO 2 : saturation artérielle en oxygène; 1,34: constante de Hüfner (en ml/g).
Le DO2 est le produit du débit cardiaque (DC) et du contenu artériel en oxygène (CaO2):
DO2 = DC × CaO2
Le DC est le produit de la fréquence cardiaque et du volume d’éjection systolique, ce dernier dépendant de la précharge cardiaque («preload»), de la contractilité (inotropisme) et de la postcharge («afterload»). Outre la fréquence cardiaque, la synchronie entre l’oreillette et le ventricule ainsi qu’entre le cœur gauche et le cœur droit est essentielle pour qu’un DC suffisant puisse être généré.
Le CaO2 est le produit de la saturation artérielle en oxygène (SaO2), du taux d’hémoglobine (Hb) et de la constante de Hüfner. Cette dernière correspond à la quantité d’oxygène qu’un gramme d’hémoglobine peut fixer, soit 1,34 ml:
CaO2 = SaO2 × Hb × 1,34
Microcirculation
L’augmentation compensatoire de l’activité du système sympathique en cas de choc entraîne une constriction des artérioles précapillaires et donc une redistribution du flux sanguin dans les organes vitaux. Les altérations microcirculatoires sont renforcées par l’activation de l’inflammation et de la coagulation, ce qui peut entraîner la formation de shunts artério-veineux impliquant une distribution inégale du flux sanguin et une perméabilité vasculaire accrue avec fuite de liquide des capillaires dans l’interstitium. Le manque d’oxygène cellulaire (hypoxie) réduit la capacité tampon pour les protons produits dans la chaîne respiratoire, ce qui provoque une acidose métabolique.
Respiration cellulaire
Une grande partie de la production d’adénosine triphosphate (ATP) de notre organisme a lieu dans la chaîne respiratoire de la membrane mitochondriale interne. Les espèces réactives de l’oxygène produites lors des inflammations peuvent causer une inhibition de la chaîne respiratoire et donc un dysfonctionnement mitochondrial [1].
En raison de l’incapacité des tissus à utiliser l’oxygène (dysoxie), il se produit un passage de la glycolyse aérobie à la glycolyse anaérobie. L’accumulation de lactate qui en résulte entraîne une hyperlactatémie. Si celle-ci s’accompagne d’une acidose métabolique (voir ci-dessus), on parle d’acidose lactique [2].
Les quatre formes de choc
Les mécanismes et certaines causes typiques des quatre types de choc (hypovolémique, obstructif, cardiogénique et distributif) sont résumés dans le tableau 1. Toutes les formes de choc ont en commun un approvisionnement insuffisant en oxygène des organes, ce qui entraîne des troubles fonctionnels. Les dysfonctionnements organiques en cas de choc sont majoritairement de nature fonctionnelle et moins structurelle, des nécroses ne surviennent que de manière isolée [3].
Remplissage cardiaque entravé: – Tamponnade péricardique – Pneumothorax sous tension
Retour veineux vers le cœur entravé: – Syndrome du compartiment abdominal
Choc cardiogénique
Inotropisme perturbé: – Infarctus du myocarde (7–10% de tous les infarctus* [4]) – Myocardite
Valvulopathies, défauts anatomiques: – Insuffisance mitrale aiguë dans le cadre d’une rupture du muscle papillaire – Communication interventriculaire
Arythmies: – Tachyarythmies – Bradycardie – Dyssynchronie entre l’oreillette et le ventricule
Choc distributif (inflammatoire)
Infection/ sepsis
Polytraumatisme
Opérations majeures
Pancréatite
Anaphylaxie
* Taux de chocs cardiogéniques chez les patientes et patients victimes d’un infarctus du myocarde avec sus-décalage du segment ST (STEMI).
Choc hypovolémique
Le choc hypovolémique est causé par une perte du volume sanguin circulant. Sur le plan physiopathologique, la diminution de la précharge cardiaque («preload») entraîne, conformément à la loi d’Otto Frank et Ernest Starling (courbe de Frank-Starling), une diminution du volume d’éjection systolique.
Choc obstructif
Le choc obstructif se caractérise par une obstruction du flux sanguin et, par conséquent, un remplissage cardiaque inadéquat. Il en résulte une diminution du volume d’éjection systolique et, donc, une réduction du DC.
L’embolie pulmonaire est responsable d’une augmentation aiguë de la postcharge du ventricule droit. De même, la postcharge du ventricule droit augmente dangereusement lorsque la pression intrathoracique est fortement accrue en raison d’un pneumothorax sous tension ou d’une auto-PEP élevée (PEP: pression expiratoire positive). Le ventricule droit, du fait de la faible épaisseur de sa paroi, ne peut que difficilement compenser ce phénomène, ce qui entraîne sa dilatation et le décalage du septum intraventriculaire vers la gauche (signe D à l’échocardiographie), de sorte que le volume télédiastolique ventriculaire gauche diminue. En cas de tamponnade péricardique, le remplissage de l’oreillette droite et/ou du ventricule droit est le plus souvent entravé, tandis qu’en cas de syndrome du compartiment abdominal, le retour veineux vers le cœur droit est perturbé.
Choc cardiogénique
En cas de choc cardiogénique, une maladie cardiaque primaire entraîne une diminution de la fonction de pompe et du DC. La diminution de la perfusion myocardique et l’ischémie qui en résultent provoquent une spirale descendante. L’activation compensatoire du système sympathique augmente la fréquence cardiaque et la postcharge et aggrave l’ischémie en augmentant le besoin en oxygène du cœur.
L’infarctus du myocarde et ses complications (par ex. rupture du septum ventriculaire, rupture du muscle papillaire, fibrillation ventriculaire) sont les causes les plus fréquentes de choc cardiogénique. Outre d’autres maladies du ventricule gauche, une insuffisance cardiaque droite aiguë peut également être un déclencheur.
Choc distributif
Les motifs moléculaires associés aux pathogènes («pathogen-associated molecular patterns», PAMPs), par exemple les lipopolysaccharides (LPS) de la paroi cellulaire des bactéries à Gram négatif, déclenchent la production de médiateurs inflammatoires. En cas de lésion tissulaire, des motifs moléculaires associés aux dégâts («damage-associated molecular patterns», DAMPs) endogènes, des activateurs du système immunitaire inné, sont libérés. Il en résulte une réponse inflammatoire avec vasodilatation et perméabilité capillaire accrue («capillary leak»), ce qui est responsable d’un retour veineux vers le cœur réduit avec diminution de la précharge cardiaque («preload») et du volume d’éjection systolique [1].
La cause classique d’un choc distributif est le sepsis. Le sepsis est un dysfonctionnement organique potentiellement fatal dû à un dérèglement de la réponse de l’organisme à une infection [5]. Le choc septique est défini par la nécessité d’un traitement vasopresseur pour maintenir une pression artérielle moyenne (PAM) ≥65 mm Hg. Les dysfonctions organiques peuvent être quantifiées par une détérioration du score SOFA («Sequential or Sepsis-related Organ Failure Assessment») de ≥2 points.
Si, au cours de la phase précoce du choc septique, le volume d’éjection systolique est réduit en raison de la diminution de la précharge cardiaque, la réponse compensatoire au stress entraîne une tachycardie et une augmentation de la contractilité [1]. Par la suite, une cardiomyopathie septique médiée par les cytokines peut provoquer un trouble réversible de la contractilité des ventricules gauche et droit [6].
Les formes non infectieuses de choc distributif surviennent souvent après une chirurgie majeure, des brûlures étendues et des polytraumatismes. En cas de choc anaphylactique, l’hypovolémie relative est induite par une libération massive d’histamine par les mastocytes et les granulocytes basophiles.
Combinaison des formes de choc
Les quatre formes de choc surviennent souvent de manière combinée dans la pratique clinique quotidienne, concomitamment et/ou l’une après l’autre. Par exemple, suite à un traumatisme, la perte de sang peut conduire à un choc hypovolémique. Une tamponnade péricardique concomitante peut provoquer un choc obstructif, tandis qu’une contusion cardiaque favorise la survenue de troubles du rythme et d’une composante cardiaque du choc. Le choc hypovolémique, obstructif et cardiogénique ainsi que les éventuelles infections nosocomiales déclenchent une inflammation, qui est souvent responsable d’un choc distributif.
Causes iatrogènes
Différentes causes iatrogènes peuvent déclencher un choc ou l’aggraver [6, 7]. Ainsi, un traitement diurétique inadéquat peut entraîner un choc hypovolémique en raison de l’excrétion accrue d’eau. Un choc cardiogénique peut également être causé par des effets indésirables de médicaments, par exemple par un surdosage de β-bloquants ou d’antagonistes calciques avec dépression myocardique consécutive.
En cas de ventilation mécanique, une expiration incomplète avec développement d’une auto-PEP peut être à l’origine d’un choc obstructif. Cette situation est reconnaissable à la courbe de débit respiratoire du respirateur. L’inspiration suivante débute alors déjà avant la fin du flux expiratoire.
Clinique et diagnostic
Evaluation du remplissage et du volume d’éjection systolique
Les veines du cou sont utilisées pour l’évaluation non invasive du remplissage intravasculaire et de la précharge cardiaque («preload»). Ces veines sont normalement remplies en position allongée et vides lorsque le tronc est relevé à 45°. En cas d’hypovolémie, les veines du cou sont vides en position allongée; en cas de choc obstructif et d’insuffisance cardiaque droite, elles sont congestionnées lorsque le tronc est surélevé à 45°. La pression veineuse centrale (PVC) peut être mesurée à l’aide d’un cathéter veineux central (CVC). La PVC (norme 5–10 mm Hg) est augmentée en cas de choc cardiogénique et obstructif, alors qu’elle est abaissée en cas de choc hypovolémique et distributif.
L’hypotension artérielle de la circulation systémique (pression artérielle systolique <95 mm Hg ou PAM <65 mm Hg) est un signe fréquent, mais non obligatoire, du choc. Une réponse adrénergique compensatoire entraîne une augmentation de la fréquence cardiaque et donc de l’indice de choc (fréquence cardiaque/pression artérielle systolique >1). La stimulation sympathique engendre une vasoconstriction et donc une augmentation de la pression diastolique, raison pour laquelle la pression moyenne peut rester longtemps normale, en particulier chez les patientes et patients jeunes.
Un cathéter artériel permet de surveiller en continu la pression artérielle, d’effectuer des gazométries artérielles répétées et de déterminer les taux de lactate. Une augmentation plate de la courbe de la pression artérielle indique une contractilité altérée. Une petite différence entre la pression artérielle diastolique et systolique peut évoquer un volume d’éjection systolique réduit et une hypovolémie. Une pression artérielle diastolique basse est un signe possible de vasoplégie.
L’échocardiographie transthoracique permet d’évaluer la fonction de pompe ventriculaire gauche et droite, ainsi que la présence d’un épanchement péricardique et de valvulopathies. Une veine cave inférieure fine avec une grande variabilité respiratoire peut témoigner d’une hypovolémie [8].
Un monitorage hémodynamique étendu est nécessaire pour déterminer le volume d’éjection systolique et le DC. Outre le cathéter artériel pulmonaire (cathéter de Swan-Ganz), le cathéter «pulse contour cardiac output» (PiCCO) représente une alternative [9]. La combinaison de la thermodilution transpulmonaire et de l’analyse du contour de l’onde de pouls artériel permet de déterminer les paramètres de la précharge cardiaque (volume télédiastolique global [VTDG]), de la contractilité (indice de fonction cardiaque [IFC]) et de la distribution de liquide dans les poumons (eau pulmonaire extravasculaire [EPEV]) [10].
Lors de l’évaluation d’un choc, il faut également être attentif à la présence éventuelle d’une hypertension intra-abdominale, déterminée par la mesure de la pression à l’aide d’une sonde vésicale. La pression intra-abdominale a des effets négatifs sur le retour veineux et la précharge cardiaque en raison de la compression des organes et du déplacement du diaphragme en direction crâniale. Elle entraîne en outre des problèmes respiratoires (de ventilation) et un dysfonctionnement des reins [11].
Signes d’hypoperfusion tissulaire
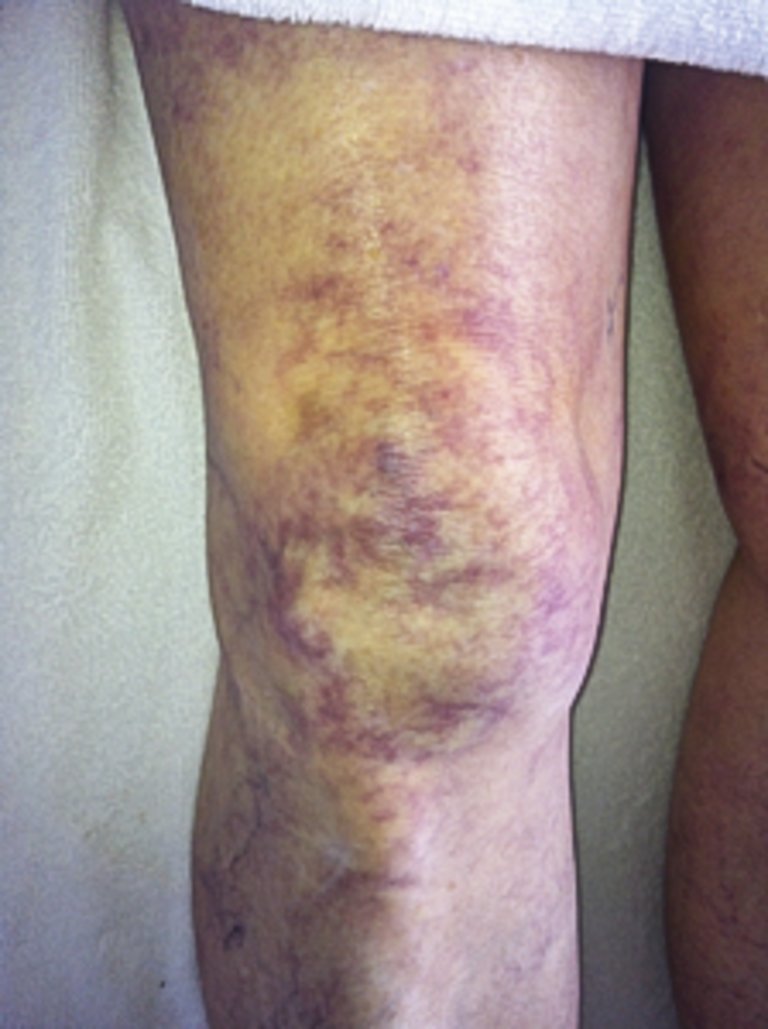
L’examen de la peau fournit des indications importantes quant à la présence d’une hypoperfusion périphérique. Une périphérie froide (accès de froid), des extrémités marbrées («mottling») (fig. 2) et un temps de remplissage capillaire prolongé lors de la pression sur le lit de l’ongle (>2 secondes) évoquent un choc.
Figure 2: Extrémités marbrées indiquant un choc (photo de patient anonyme, prise par A. Rudiger).
Une hyperlactatémie révélée par la gazométrie artérielle est un signe d’alarme et indique un déficit énergétique au niveau cellulaire. Il est considéré que le mécanisme sous-jacent est l’hypoxie tissulaire avec glycolyse anaérobie. La libération de lactate à partir des muscles, stimulée par les sympathicomimétiques, augmente les taux de lactate dans le sang; en parallèle, une dysfonction hépatique concomitante peut entraver l’élimination du lactate (tab. 2) [2, 8, 12]. Normalement, les taux de lactate dans le sang sont d’environ 1 mmol/l et, en cas de choc, ils augmentent jusqu’à >2,2 mmol/l [8, 13].
Tableau 2: Diagnostics différentiels de l’hyperlactatémie.
Métabolisme anaérobie en cas d’hypoxie tissulaire
Choc
Effort physique intense, y compris crise d’épilepsie de type «grand mal»
Médicaments/intoxications
β-agonistes
Alcool
Metformine
Médicaments antirétroviraux
Paracétamol
Dysfonction hépatique
Hyperglycémie, acidocétose diabétique
Leucémies et lymphomes
Un autre signe d’hypoperfusion tissulaire est la chute de la saturation veineuse centrale en oxygène dans le sang cave supérieur (ScvO2) [13]. En cas de DC diminué, d’anémie et d’hypoxémie, celle-ci est réduite en raison de l’augmentation de l’extraction d’oxygène en périphérie. Dans le choc distributif, la ScvO2 est basse au cours de la phase précoce [14], se normalise avec la mise en œuvre d’une expansion volémique et est pathologiquement élevée chez les patientes et patients dont l’extraction de l’oxygène est perturbée (en raison de troubles microcirculatoires et d’un dysfonctionnement mitochondrial) [8].
Dysfonctionnements organiques en cas de choc
L’hypoperfusion cérébrale se traduit par des troubles de la conscience qualitatifs et quantitatifs.
Du fait du lien étroit entre la perfusion sanguine rénale et la filtration glomérulaire, une insuffisance rénale aiguë peut survenir en raison d’une hypoperfusion dans le cadre d’un choc. La sévérité de l’insuffisance rénale aiguë peut être quantifiée à l’aide de la classification en stades de la «Kidney Disease: Improving Global Outcomes» (KDIGO) (tab. 3). Cette classification se base sur une diminution de l’excrétion urinaire (oligurie à anurie), qui se manifeste en l’espace de quelques heures en cas de choc, ainsi que sur une augmentation du taux de créatinine sérique, ce qui prend plusieurs jours. Outre la forme prérénale de l’insuffisance rénale à la phase précoce du choc, une forme rénale d’insuffisance rénale médiée par les cytokines apparaît secondairement. Cela explique pourquoi l’insuffisance rénale persiste pendant des jours, voire des semaines, chez de nombreux patients et patientes, même lorsque la pression de perfusion et le DC ont été corrigés par l’administration de liquides et de vasopresseurs.
Tableau 3: Classification en stades de l’insuffisance rénale aiguë (KDIGO 2012 [15]).
Stade
Créatinine sérique
Excrétion urinaire
1
Augmentation de 1,5–1,9 fois ou Augmentation ≥26,5 µmol/l
<0,5 ml/kg/h sur 6–12 h
2
Augmentation de 2–3 fois
<0,5 ml/kg/h sur ≥12 h
3
Augmentation de ≥3 fois ou Augmentation ≥354 µmol/l ou Début d’une thérapie de substitution rénale
<0,3 ml/kg/h sur ≥24 h ou Anurie sur ≥12 h
KIDGO: «Kidney Disease: Improving Global Outcomes»; h: heure (n).
Outre le cerveau et les reins, le foie réagit également à une diminution de la perfusion. En cas de choc cardiogénique, la diminution de la perfusion via l’artère hépatique et la rétention sanguine dans la circulation hépatique entraînent une hépatite hypoxique avec augmentation des transaminases sériques [16]. Les malades en état critique présentent souvent une augmentation de la bilirubine, dont l’origine est multifactorielle [17].
L’encéphalopathie («Glasgow Coma Scale» [GCS]), l’insuffisance rénale aiguë (taux de créatinine, excrétion urinaire) et la dysfonction hépatique (taux de bilirubine) font partie du score SOFA, qui est corrélé à la mortalité des patientes et patients en soins intensifs [18].
Le tableau 4 résume quelques paramètres cliniques, hémodynamiques et de laboratoire typiques de l’hypoperfusion.
Dysfonction hépatique: augmentation de l’alanine aminotransférase (ALT), de l’aspartate aminotransférase (AST) et de la bilirubine
L’essentiel pour la pratique
• Les quatre formes de choc – hypovolémique, obstructif, cardiogénique et distributif – peuvent survenir de manière isolée, combinée ou successive.
• Les troubles microcirculatoires cutanés, l’hypotension artérielle, la tachycardie et l’encéphalopathie sont des signes cliniques majeurs.
• L’hyperlactatémie et l’acidose métabolique détectées par la gazométrie artérielle renforcent la suspicion de choc.
• Il convient de songer à la possibilité d’influences iatrogènes.
Disclosure statement
Les auteurs ont déclaré ne pas avoir de conflits d’intérêts potentiels.
Correspondance
Dr méd. Johann Stuby Institut für Anästhesiologie Universitätsspital Zürich Rämistrasse 100 CH-8091 Zürich johannstuby[at]aol.com
Références
1 Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis-induced myocardial dysfunction: pathophysiology and management. Journal of intensive care. 2016;4(1):22.
2 Kraut JA, Madias NE. Lactic acidosis. New England Journal of Medicine. 2014;371(24):2309–19.
3 Rudiger A, Stotz M, Singer M. Cellular processes in sepsis. Swiss Medical Weekly. 2008;138(43–44):629–34.
4 Kolte D, Khera S, Aronow WS, Mujib M, Palaniswamy C, Sule S, et al. Trends in Incidence, Management, and Outcomes of Cardiogenic Shock Complicating ST-Elevation Myocardial Infarction in the U nited S tates. Journal of the American Heart Association. 2014;3(1):e000590.
5 Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–10.
6 Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Critical care medicine. 2007;35(6):1599–608.
7 Singer M, Glynne P. Treating critical illness: the importance of first doing no harm. PLoS medicine. 2005;2(6).
8 Vincent J-L, De Backer D. Circulatory shock. New England Journal of Medicine. 2013;369(18):1726–34.
9 Ritter S, Rudiger A, Maggiorini M. Transpulmonary thermodilution-derived cardiac function index identifies cardiac dysfunction in acute heart failure and septic patients: an observational study. Critical Care. 2009;13(4):R133.
10 Gassanov N, Caglayan E, Nia A, Erdmann E, Er F. Der PiCCO-Katheter. DMW-Deutsche Medizinische Wochenschrift. 2010;135(46):2311–4.
11 Malbrain ML, De Waele J, De Keulenaer BL. What every ICU clinician needs to know about the cardiovascular effects caused by abdominal hypertension. Anaesthesiology intensive therapy. 2015;47(4):388–99.
12 Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Annals of intensive care. 2013;3(1):1–8.
13 Hauffe T, Krüger B, Bettex D, Rudiger A. Shock management for cardio-surgical ICU patients–the golden hours. Cardiac failure review. 2015;1(2):75.
14 Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. New England Journal of Medicine. 2001;345(19):1368–77.
15 Kellum JA, Lameire N, Aspelin P, Barsoum RS, Burdmann EA, Goldstein SL, et al. Kidney disease: improving global outcomes (KDIGO) acute kidney injury work group. KDIGO clinical practice guideline for acute kidney injury. Kidney international supplements. 2012;2(1):1–138.
16 Nikolaou M, Parissis J, Yilmaz MB, Seronde M-F, Kivikko M, Laribi S, et al. Liver function abnormalities, clinical profile, and outcome in acute decompensated heart failure. European Heart Journal. 2013;34(10):742–9.
17 Geier A, Fickert P, Trauner M. Mechanisms of disease: mechanisms and clinical implications of cholestasis in sepsis. Nature clinical practice Gastroenterology & Hepatology. 2006;3(10):574–85.
18 Jain A, Palta S, Saroa R, Palta A, Sama S, Gombar S. Sequential organ failure assessment scoring and prediction of patient’s outcome in Intensive Care Unit of a tertiary care hospital. Journal of Anaesthesiology, Clinical Pharmacology. 2016;32(3):364.