

Publié le 03.08.2021
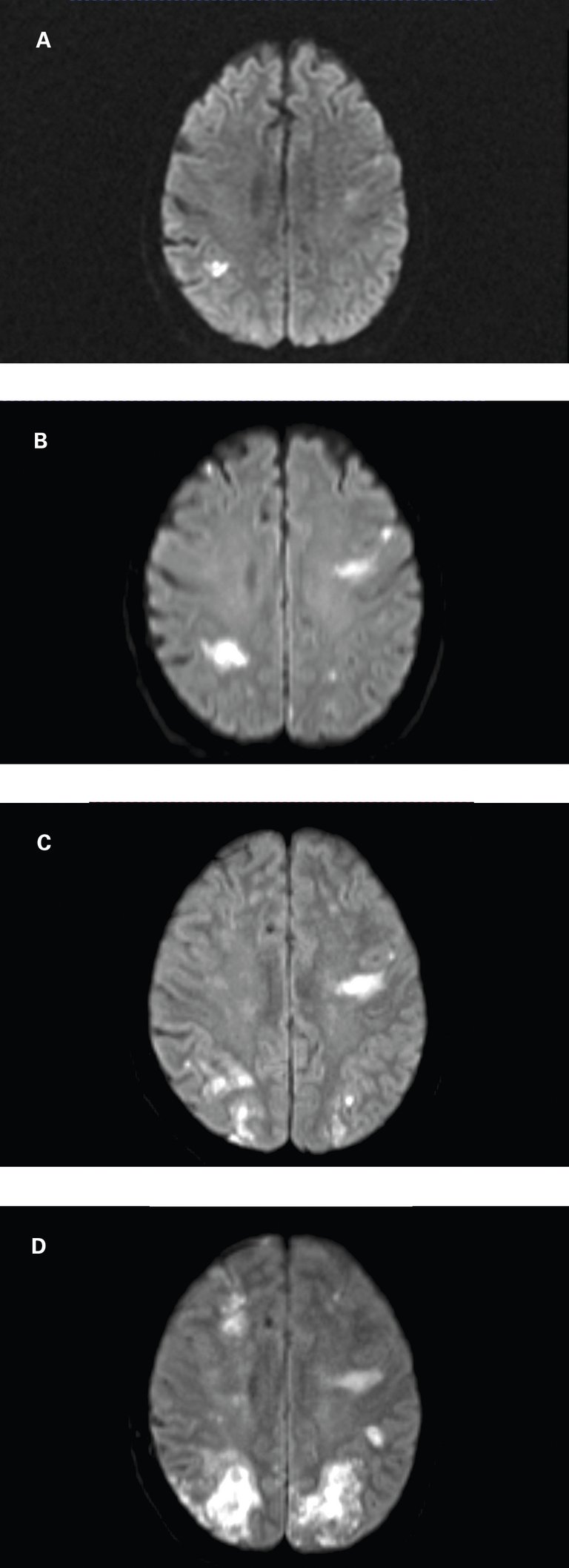
Une patiente de 71 ans nous a été adressée en urgence en raison d’une détérioration neurologique aiguë, après avoir été hospitalisée à deux reprises au cours des deux derniers mois en raison d’ischémies cérébrales.
| Tableau 1: Causes d’ischémies cérébro-vasculaires multiples. |
| Embolies cardiaques ou artérielles |
| Thrombose veineuse et thrombose des sinus veineux cérébraux |
| Maladies inflammatoires dues à un agent pathogène, par ex. vascularite associée au virus varicelle-zona |
| Maladies inflammatoires auto-immunes, entre autres angéite primitive du système nerveux central (APSNC) |
| Angiopathie amyloïde cérébrale (inflammatoire) |
| Syndrome de vasoconstriction cérébrale réversible (SVCR) |
| Cause paranéoplasique |
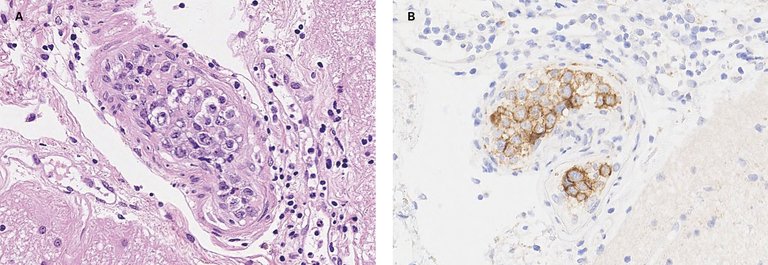
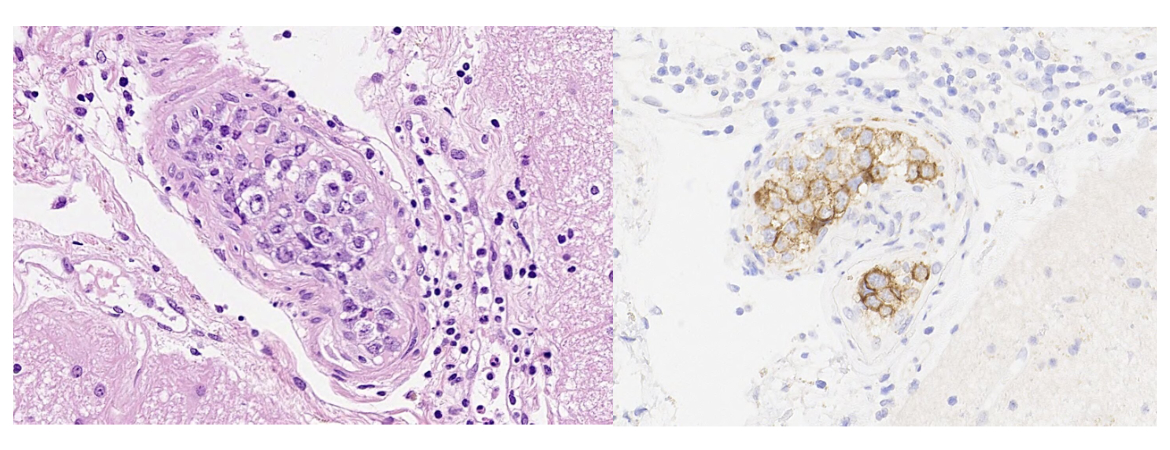
Publié sous la licence du droit d'auteur.
"Attribution - Non-Commercial - NoDerivatives 4.0"
Pas de réutilisation commerciale sans autorisation..
See: emh.ch/en/emh/rights-and-licences/
